















 30 / 38
30 / 38

J u n e 1 1 - 1 3 , 2 0 1 8 | D u b l i n , I r e l a n d
allied
academies
Page 54
CANCER STEM CELLS AND
ONCOLOGY RESEARCH
11
th
International Conference on
Journal of Medical Oncology and Therapeutics
|
Volume 3
Miaofen G Hu et al., J Med Oncl Ther 2018, Volume 3
CDK6-MEDIATED SUPPRESSION
OF CD25 IS REQUIRED FOR SELF-
RENEWAL OF LSCS
Miaofen G Hu
1
, Nilamani Jena
1, 2
, Alexander J Hu
1, 3
, Wei Li
1
,
Jamie K Hu
1, 4
and
Richard Van Etten
1,2
1
Tufts Medical Center, USA
2
University of California Irvine, USA
3
Tufts University School of Medicine, USA
4
Yale School of Medicine, USA
D
espite recent advances in chemotherapy, relapse is frequent, possibly
because the available therapies do not eradicate the cells that initiate
and sustain the disease
in vivo
, so-called leukemia stem cells (LSCs).
Cyclin-dependent kinase 6 (CDK6) regulates cell cycle progression and
modulates differentiation of certain cells. It is predominantly expressed
in hematopoietic cells and over-expressed in human T-ALL/LBL. To
clarify the role of CDK6 in cell cycle control and tumorigenesis, I have
generated mice with targeted mutations in Cdk6. These “knock-in” alleles
generate hyperactive or inactive kinase subunits that may better mimic
hyperactivation of CDK6 in tumor cells or model pharmaceutical inhibition
of CDK6, respectively. We have found that CDK6 is required for initiation
and maintenance of T-ALL leukemia and lymphomagenesis induced by
constitutively active Notch/Myr-AKT. Pharmacologic inhibition of CDK6
kinase induces CD25 expression, cell cycle arrest, and apoptosis inmouse
and human T-ALL. Ablation of Cd25 in a K43M background restores
Notch-induced T-leukemogenesis, with disease that is resistant to CDK6
inhibitors
in vivo
. Moreover, loss of Cd25 in a K43M background restore
the ability of LSCs to self-renew. These data support a model whereby
CDK6-mediated suppression of CD25 is required for initiation of T-ALL by
activated Notch1, and CD25 induction mediates the therapeutic response
to CDK6 inhibition in established T-ALL. These results both validate CDK6
as a molecular target for therapy
of this subset of T-ALL and
suggest that CD25 expression
could serve as a biomarker for
responsiveness of T-ALL to
CDK4/6 inhibitor therapy.
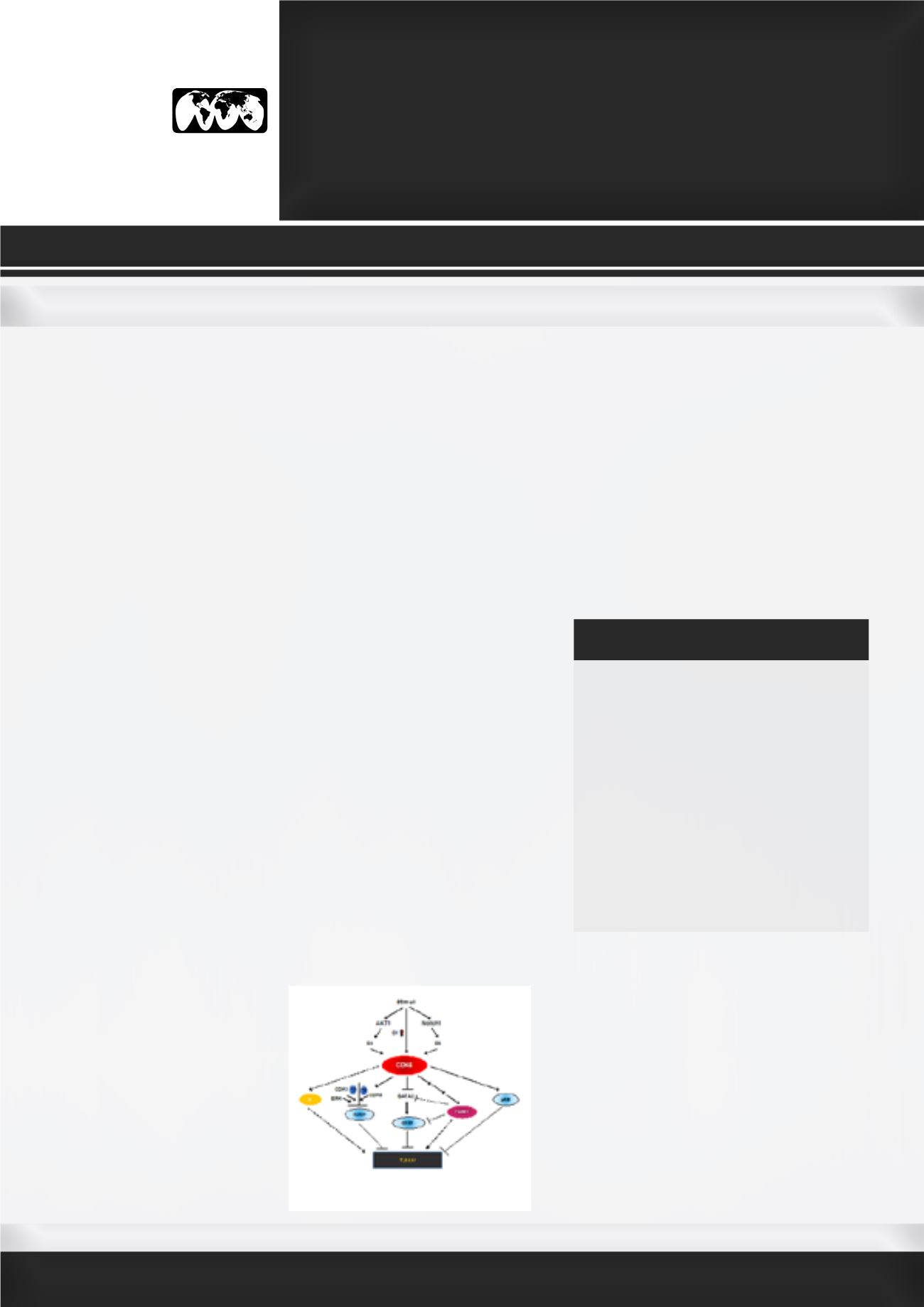
Figure. A working model of the role of
CDK6 in T-ALL. In response to stimuli,
increased expression of cyclin D1
and CDK6 leads to increased CDK6
activation, while Notch1 and AKT1
are also independently activated in
parallel. Notch1 further activates CDK6
via upregulation of CDK6 and/or by
Miaofen G Hu has completed her PhD from
Boston University School of Medicine and post-
doctoral studies fromHarvard University School
of Medicine. She has been Assistant Professor
at TUFTS medical Center since 2011. She has
published 24 papers in reputed journals. Her
most significant research accomplishments
thus far include creating a CDK6 mouse model,
discovering the role of CDK6 as a common me-
diator of Notch1 and AKT1 signaling pathways,
establishing the potential therapeutic role of
CDK6 in T cell malignance, revealing the func-
tion of CDK6 kinase activity in negatively reg-
ulating the conversion of fat-storing cells into
fat-burning cells.
mhu@tuftsmedicalcenter.orgBIOGRAPHY
increased cyclin D3 protein, while AKT1 activates CDK6
through the stabilization of cyclin D2. Once activated,
CDK6 can phosphorylate pRB, resulting in its inactivation.
CDK6, along with ERK and CDK1 can also phosphorylate
RUNX1 thereby promotingRUNX1proteolyticdegradation.
On a different molecular path, phosphorylation of FOXM1
by CDK6 stabilizes FOXM1, which in turn promotes
methylation of the GATA3 promoter, decreasing GATA3
expression and the subsequent recruitment to the
CD25 proximal promoter region. CD25 expression is
consequently reduced and T-ALL develops. Devoid of
CDK6 protein/kinase activity, pRB and RUNX1 remain
active, which suppress the tumorigenesis in CD25-
independent manner. Contrastingly, without CDK6, FOXM1
is in its inactive state, leading to CD25 upregulation.
Overall, T-ALL is suppressed by FOXM1 inactivity, and by
increasing RUNX1, pRB, GATA3, and CD25 expression or
activity.