Review Article - Journal of Chemical Technology and Applications (2018) Volume 2, Issue 1
Theoretical investigation of the mechanism, stereoselectivity and regioselectivity of the epoxidation reaction of ó-himachalene: MEDT screening.
- *Corresponding Author:
- Abdellah Zeroual
Laboratory of Physical Chemistry, Chouaïb Doukkali University, P.O. Box 20, 24000, El Jadida, Morocco.
Tel: +212667845404
E-mail: zeroual19@yahoo.fr
Accepted date: February 22, 2018
Citation: El-Ghozlani M, El-Haib A, El-Ajlaoui R, et al. Theoretical investigation of the mechanism, stereoselectivity and regioselectivity of the epoxidation reaction of γ-himachalene: MEDT screening. J Chem Tech App. 2018;2(1):4-9.
Abstract
In this paper we employed the MEDT method to examinethe mechanism, stereoselectivity and regioselectivity of the epoxidation reaction of γ-himachaleneyielding a γ-epoxyhimachalene P-1,which participates in two competitive reaction channels, from the energies, IRC and density maps of the transition state we can concluded that in oxidation reaction, the m-CPBA will participated as strong electrophile via a one-step mechanism. The regioselectivity experimentally obtained was verified by the nucleophilic Parr functions. Side selectivity, namely α and β of the double bond C7=C8 was predicted using transition state theory. This stereoselectivity is kinetically controlled in good conformity with experimental outcomes.
Keywords
γ-himachalene, Regioselectivity, m-CPBA, Stereoselectivity, DFT, MEDT.
Introduction
Plants represent an immense source of complex chemical molecules exploited in the perfume, agro-food, cosmetic and pharmaceutical industries. Most plants contain essential oils; they are then entitled “aromatic plants". These essential oils are found in many parts of the plant: wood, leaves, fruits, bark, seeds and roots. These are complex mixtures consisting of several tens, or even more than one hundred compounds, mainly terpenes and aromatic compounds. Essential oils have many biological activities; they are utilized for their antiseptic properties against bacterial diseases, for example against the endocranial bacteria [1] or at the level of the vaginal microflora [2,3]. However, they also have cytotoxic properties [4] that bring them closer to antiseptics and disinfectants as broad spectrum antimicrobial agents [5]. The essential oils have antibacterial properties, antioxidant activity [6] and potential applications in foods [7], essential oils or their active compounds could also be used as protection agents against phytopathogenic fungi and microorganisms invading foodstuffs [8,9].The essential oils of cedar of the Atlas which constituted by three elements such as α-himachalene, β-himachalene and γ-himachalene (Scheme 1 and Figure 1).

Figure 1: B3LYP/6-31G(d) 3D maps of the orbital HOMO and LUMO of the γ-himachalene and m-CPBA.

Scheme 1: Structure of α-himachalene, β-himachalene and γ-himachalene.
To improve the chemical and biological proprieties, the γ-himachalene, treated at ambient temperature with a stoichiometric quantity of m-chloroperbenzoic acid (m-CPBA) in chloroform, Regio specifically and stereospecifically leads to γ-epoxyhimachalene P-1 in quantitative yield (Scheme 2).

Scheme 2: epoxidation reaction of γ-himachalene.
Herein, in order to comprehend the stereo-selectivity, the regioselectivity and the molecular mechanism of the epoxidation reaction of the γ-himachalene is performed in the MEDT using DFT/B3LYP/6-31G (d) methods.
Methods of Calculation
All geometry optimizations computation was executed using the Gaussian 09 programs [10]. The geometries of the products were fully optimized through DFT calculations using the B3LYP functional [11,12] jointly in addition to the 6-31G(d) basis set [13]. The transition states, resultant to the two α and β epoxidation sides, were located at the B3LYP/6-31G (d) level by QST2 and QST3, their existence was validated by the existence of one and only one imaginary frequency in the Hessian matrix. The IRC [11] was performed and plotted to show that the TS is well connected to both minima (reagents and product). The effects of dichloromethane in energy calculations were taken into account using the model of the polarizable continuum (PCM) developed by the Tomasi group [14,15] as part of the self-consistent reaction field (SCRF) [16-18] Values of energies in dichloromethane were calculated with the standard statistical thermodynamics at 258 C and 1 atm. The total electron density transfer (GEDT) was calculated via the amount of the natural atomic charges (q), found by a natural population analysis (NPA). The global electrophilicity index, [19-21] w, is granted by the following formula, 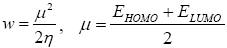 and
and 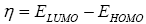 are the electronic chemical potential and the chemical hardness respectively [22,23]. The nucleophilicity index, [24-26] N is defined as
are the electronic chemical potential and the chemical hardness respectively [22,23]. The nucleophilicity index, [24-26] N is defined as 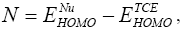 Electrophylic
Electrophylic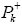 and nucleophilic
and nucleophilic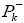 Parr functions, [27-54] were reached by the analysis of Mulliken's atomic spin density.
Parr functions, [27-54] were reached by the analysis of Mulliken's atomic spin density.
Results and Discussion
This part has been divided in two sections: (1) Analyze of the global DFT indices of reagent ofthe epoxidation reaction of γ-himachalene to understand the regioselectivity observed. (2) Next, Relative Gibbs free energy for the stationary points of the epoxidation reaction of γ-himachalene are explored and analyzed.
Analyze of the global indexes of reagents
The indexes specifically, μ the electronic chemical potential, η chemical hadrness, ω electrophilicity and N nucleophilicity, are summarized in Table 1 and 3D maps of the orbital HOMO and LUMO of the γ-himachalene and m-CPBA are represented in Figure 2.
| Compunds | η | µ | ω | N |
|---|---|---|---|---|
| γ-himachalene | 6.61 | -2.56 | 0.49 | 3.65 |
| m-CPBA | 5.29 | -4.35 | 1.79 | 2.52 |
Table 1: B3LYP/6-31(d) electronic chemical potential μ, chemical hadrness η, electrophilicity ω and nucleophilicity N in ev of the γ-himachalene and m-CPBA.

Figure 2: B3LYP/6-31G(d) 3D maps of the orbital HOMO, electrostatic potential on the 0.001andnucleophilic Parr function of γ-himachalene.
The electronic chemical potential of γ-himachalene, μ=−2.56 eV, is elevated than that of m-CPBA, μ=−4.35 eV, thus signifying that the global electron density transfer (GEDT) will go from γ-himachalene towards m-CPBA.
The m-CPBA presents an electrophilicy ω index of 1.79 eV and a nucleophilicity N index of 2.52 eV, being classified as strong electrophile than ω index of the γ-himachalene, 0.43 eV and a nucleophilicity N index of 3.65 eV. Consequently, m-CPBA is classified as a strong electrophile, while the γ-himachalene a strong nucleophile. On the other hand, we can observe from the Figure 1 that, ΔE1=4.16 smaller than ΔE2=7.74 (eV) confirming that the global electron density transfer will go from γ-himachalene towards m-CPBA.
Prediction of the regioselectivity of the reaction using local indices
It has been established that along a polar reaction with asymmetric reagents the most favorable reactive channel is that where the two-centre interaction is developed between the most electrophilic centre of the electrophile and the most nucleophilic centre of the nucleophile. The electrophilic and the nucleophilic Parr functions have been reported as derived from the changes of spin electron density attained by a GEDT process developed from the nucleophile towards the electrophile. According to the Parr functions, the most favorable single bond formation arises between the most electrophilic and nucleophilic center of the reagents, the electrostatic potential is a true character, a physical noticeable. It can be found computationally. Even as it has been employed to understand, and predicting the regioselectivity. Regions where V(r)>0 can be expected to be attracted favorably, at least initially, to negative segment of other molecules, while V(r)<0 predicts attractive interactions with positive segment. In this part, we obtain V(r) with the density functional B3LYP/6- 31(d) (Figure 2).
We represent in Figure 2, the isodensity of the HOMO molecular orbital, the electrostatic potential and the nucleophilic Parr functions of γ-himachalene.
The representation of the iso-density map of the HOMO molecular orbital of γ -himachalene (Figure 2a), shows that the HOMO orbital is highly condensed at the C7=C8 double bond, so the attack of an equivalent of m-CPBA is preferentially in this double bond.
We can observed from Figures 2b and 2c that surface map value of the bond C2=C3 and C7=C8 are -0.016 and -0.019 respectively, indicating that the double bond C7=C8 is very nucleophilic than anther double bond, the fact that this reaction was regioselective, in addition an analysis of the nucleophilicp_ k^- Parr functions at the reactive sites of γ-himachalene indicates that the C7 and C8 carbon atoms, with P_k^- value of 0.18 and 0.28 respectively, are more nucleophilically activate than the C2 and C3 carbon atoms, with a P_k^- value of 0.16 and 0.19 respectively.Consequently, the attack is preferentially effected on the double bond C7=c8 in good agreement with experimental observations.
Energetic study of the two modes of attack
Due to the asymmetry of the γ-himachalene, we have two stereoisomeric reactive channels (Scheme 3) depending on the relative position of the double bond C7=C8 (α-side and β-side). The values of the free energy and the relative free energy ones of the stationary points involved in the in-epoxidation reaction of γ-himachalene by m-CPBA are summarized in Table 2, the energy profile of this epoxidationis given in Figure 3 and the density map of the transition is given in Figure 4.
| Variables | G | ΔG | Δ(ΔG) |
|---|---|---|---|
| Reagents | -1541.15247 | ------- | ------- |
| TS-1 | -1541.08798 | 40.47 | 48.17 |
| TS-2 | -1540.91121 | 88.64 | |
| P-1+m-CPBA | -1541.18646 | -21.32 | 1.12 |
| P-2+m-CPBA | -1541.18825 | -22.44 |
Table 2: B3LYP/6-31G(d) total (G, in a.u) and relative an energies (ΔG, in kcal mol−1) in dichloromethane of the stationary points involved in epoxidation reaction of γ-himachalene by m-CPBA.

Figure 3: B3LYP/6-31G(d) Gibbs free energy profile (ΔG, in Kcal/ mol) of the epoxidation reaction of the γ-himachalene.

Figure 4: the density map of the transition state of the α-side and β-side of the double bond C7=C8.

Scheme 3: Studied competitive stereoisomeric channels associated with epoxidation reaction of γ-himachalene.
The activation energy associated with the electrophilic attack of the O2 oxygen of m-CPBA on the α-side of the double bond of γ-himachalene presents a high value of 88.64 kcal/mol (TS- 2), formation of the P-2 beings exothermic by -22.44 kcal/mol, the activation energy value decreases to 40.47 kcal/mol (TS-1) and formation of the P-1 beings exothermic by -21.32 kcal/mol consequently the product P-1 is kinetically favored.
The density map of the transition state indicating that the formation of the products P-2 not favored, because the density map of the reagents is separate, in opposite the density map of TS-1, γ-himachalene and oxygen of the m-CPBA are united in a density map, which indicates that formation of the products P-1 is favored.
Conclusion
The epoxidation reaction of γ-himachalene has been investigated within the DFT calculations at the 6-31(d) computational level. The analysis of the nucleophilic Parr functions of γ-himachalene indicates that the most interaction take place between double bond C7=C8 and O2 oxygen atom of the m-CPBA. An examination of the values of the energy G and the relative energy ΔG of this epoxidation show that it takes place through a one-step mechanism. The exothermic nature of epoxidation reaction makes the formation of the two stereoisomiric products P-1 and P-2 irreversible and in conclusion α-side position (P- 1) is kinetically favored in good agreement with experimental result.
References
- Pellecuer J, Jacob M, Buochberg MS de, et al.Tests on the use of essential oils from aromatic mediterranean plants in preservative odontology. PlantesMédicinales et Phytothérapie.1980;14:83-98.
- Violon C, Leger D, Chaumont JP. Activités antagonistes, in vitro, de certains composés volatils naturels vis-à-vis de germes de la flore vaginale. Plantes Médicinales et Phytothérapie.1993;26:17-22.
- Chaumont JP, Le´ger D. Proprie´te´s antifongiques de quelques phe´nols et de compose´s chimiquement voisins, relation structureactivite. Plant Med Phytother.1989;23:124-28.
- Sivropoulou A, Papanikolaou E, Nikolaou C, et al. Antimicrobial and cytotoxicactivities of origanum essential oils. J AgricFood Chem.1996;44:120-05.
- Andogan BC, Baydar H, Kaya S, et al. Antimicrobial activity and chemical composition of some essential oils. Archives of pharmacal research.2002;25:860-64.
- Karioti A, Hadjipavlou-Litina D, Mensah ML, et al. Composition and antioxidant activity of the essential oils of Xylopia aethiopica (Dun) A. Rich. (Annonaceae) leaves, stem bark, root bark, and fresh and dried fruits, growing in Ghana. J Agric Food Chem.2004;52:8094-8.
- Burt S. Essential oils: their antibacterial properties and potential applications in foods-a review. Int J Food Microbiol.2004;94:223-53.
- Okigbo RN, Anuagasi CL, Amadi JE. Advances in selected medicinal and aromatic plants indigenous to Africa.J Med Plant Res.2009;3:86-95.
- McGaw LJ, Eloff JN. Ethnoveterinary use of southern African plants and scientific evaluation of their medicinal properties. J Ethnopharmacol. 2008;119:559-74.
- Frisch J, Trucks GW, Schlegel HB, et al.Gaussian 09, Revision a.1, Gaussian, Inc., Wallingford, CT 2009.
- Becke AD. A new mixing of Hartree-Fock and local density-functional theories. J Chem Phys.1993;98:1372-77.
- Lee CT, Yang WT, Parr RG. Density-functional exchange-energy approximation with correct asymptotic behavior. Physical Review B.1988;37:785-89.
- Hehre WJ, Radom L, Schleyer PVR. Ab Initio Molecular Orbital Theory.Wiley, New York, USA.1986.
- Tomasi J, Persico M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem Rev.1994;94:2027-94.
- Simkin BY, Sheikhet I. Quantum chemical and statistical theory of solutions? computational approach, Ellis Horwood, London.1995.
- Cances E, Mennucci B, Tomasi J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys.1997;107:3032-41.
- Cossi M, Barone V, Cammi R, et al.Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem Phys Lett. 1997;255:327-35.
- Barone V, Cossi M, Tomasi J. Geometry optimization of molecular structures in solution by the polarizable continuum model.J Comput Chem.1998;19:404-17.
- Reed AE, Weinstock RB, Weinhold F. Natural population analysis. J Chem Phys.1998;83:735-46.
- Reed AE, Curtiss LA, Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev.1988;88:899-926.
- Parr RG, Szentpaly LV, Liu S. Electrophilicity index. J Am Chem Soc.1999;121:1922-24.
- Parr RG, Pearson RG. Absolute hardness: Comparison parameter to absolute electronegativity.J Am Chem Soc.1983;105:7512-16.
- Parr RG, Yang W. Density functional theory of atoms and molecules, Oxford University Press, New York, USA. 1989.
- Domingo LR, Chamorro E, Pérez P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J Org Chem. 73: 4615-24.
- Domingo LR, Pérez P. The nucleophilicity N index in organic chemistry. Org Biomol Chem. 2011;9:7168-75.
- Kohn W, Sham L. Self-consistent equations including exchange and correlation effects. J Phys Rev.1965;140:1133-37.
- Noury S, Krokidis K, Fuster F. Computational tools for the electron localization function topological analysis.Comput. Chem.1965;23:597-604.
- El-Haib A, El-Ajlaoui R, El-Idrissi M, et al. The mechanism, the chemoselectivity and the regioselectivity of the 1-Benzyl-4-ethynyl-1H-[1,2,3]triazole and 1-Azidomethyl-4-tert-butyl-benzene in [3+2] cycloaddition reactions: a DFT study.Mor J Chem.2018;6:14-21.
- Ourhriss N, Zeroual A, Gadhi CA, et al. Synthesis, spectroscopic NMR and theoretical (HF and DFT) investigation of 3,5,5,9-tetramethyl-2-nitro-6,7,8,9-tetrahydro-5H-benzocycloheptene and 2,5,9,9-tetramethyl-1,3-dinitro-6,7,8,9-tetrahydro-5H-benzocycloheptene.Eur J Mol Biotechnol.2017;5:52-59.
- El-Idrissi M, Zeroual A, El-Haib A, et al. Theoretical study of the mechanism and regioselectivity of electrophilic substitution reaction between the ar-himachalene and acetic anhydride?.Int J Multidisciplinary Sci.2017;2:1-10.
- El Idrissi M, Zoubir M, Zeroual A. Understanding the mechanism and regioselectivity of Prop-2-yn-1-ol with azido-compounds in [3+2] cycloaddition reactions: a molecular electron density theory study. J Marocain de ChimieHétérocyclique.2017;16:179-185.
- Zeroual A, El Idrissi M, Zoubir M, et al. Theoretical study of the mechanism and regioselectivity of Prop-2-Yn-1-Ol with Azide in [3+2] cycloaddition reactions. Open Access J Translational Med Res.2017;1:1-5.
- Zoubir M, El Idrissi M, El Ajlaoui R, et al. Theoretical study of the chemo and the regioselectivity of the Baeyer-Villiger reaction of bicyclo[3.2.0]hept-2-en-6-one by hydrogen peroxide. Euro Rev Chem Res.2017;4:28-33.
- Zeroual A, El Idrissi M, Zoubir M, et al. Theoretical study of the reactivity and regioselectivity of the addition reaction between HCl and alkenes, investigation of the Markovnikov's rule. Euro Rev Chem Res. 2017;4:21-27.
- Zeroual A, El Idrissi M, El Ajlaoui R, et al. MEDT study of the mechanism and regioselectivity of diazocompounds and alkenes in [3+2] cycloaddition reaction?. Euro J MolBiotechnol.2017;5:43-49.
- El Idrissi M, El Ajlaoui R, Zoubir M, et al. Theoretical study of the chemo- and regioselectivity of the [3+2] cycloaddition reaction between mesitonitrile oxides and 2-fluoren-9-ylidene-malononitrile. J Mater Environ Sci. 2017;8:3564-69.
- Zeroual A, Zoubir M, El Idrissi M, et al. Theoretical analysis of reactivity and regioselectivity in [1+2] cyloaddtion reaction of limonene, terpinolene and ?-terpinene with dichlorocarbene, Global Journal of Science Frontier Research: B.Chemistry.2017.
- Zoubir M, Zeroual A, El Idrissi M, et al. Experimental and theoretical analysis of the reactivity and regioselectivity in esterification reactions of diterpenes (totaradiol, totaratriol, hinikione and totarolone), Medi J Chem.6:98-107.
- Ourhriss N, Zeroual A, Ait El Had M, et al. Synthesis of 1-isopropyl-4,7-dimethyl-3-nitronaphthalene: An experimental and theoretical study of regiospecific nitration. J Mater Environ Sci.2017;8:1385-90.
- Zoubir M, Zeroual A, El Idrissi M, et al. Understanding the chemoselectivity and stereoselectivity in Michael addition reactions of ß-hydroxyparthenolides and amines such as pyrrolidine, morpholine, piperidine and 1-methylpiperazine: a DFT study. J Mater Environ Sci. 2017;8:990-96.
- Zoubir M, Zeroual A, Benharref A, et al. Understanding the Holleman Rule in the electrophilic substitution reaction using parr functions. J Comput Methods Mol Design.2016;6:1-4.
- El Idrissi M, Zoubir M, Zeroual A, et al. Theoretical study of the mechanism and regioselectivity of the 1,3-dipolar cycloaddition reaction of azides with alkynes. J=Marocain de ChimieHétérocyclique. 2017;15:146-151.
- El Idrissi M, El Haib A, Zoubir M, et al. Understanding the regioselectivite of the Baeyer-Villiger reaction of bicyclo[4.2.0]octan-7-one and bicyclo[3.2.0]heptan-6-one: A DFT Study. Journal of Computational Methods in Molecular Design. 2016;6:75-79.
- Zeroual A, Hammal R, El Hajbi A. A DFT Study of the [1+2] Cycloaddition Reactions of 2-[1,3]Dioxolan-2-ylidene-malononitrile, TCE and chlorocarbene. J Comput Methods Mol Design. 2015;5:97-101.
- Zeroual A, El Hajbi A. Understanding the regioselective and molecular mechanism of the TCE in cycloaddtion reaction (TCE+Cp) and addition reaction (TCE+HCl) using DFT calculation. Canadian Chemical Transactions.2015;3:430-437.
- Zeroual A, Barhoumi A, Bakkas S, et al. Understanding, which oxygen attacks bromotrimethylsilane in McKenna Reaction using DFT calculation? J Comput Methods Molecular Design.2015;5:150-54.
- Zeroual A, El Haib A, Benharref A, et al. A combined experimental and theoretical study of highly chemioselectivity acetylation of diterpene. J Comput Methods Molecular Design.2015;5:58-62.
- Barhoumi A, Zeroual A, Bakkas S, et al. Theoretical study of the regioselectivity of the reaction between tetrachloromethane and triethyl phosphite using the DFT B3LYP/6-31G (d) method?.J Comput Methods Molecular Design.2015;5:8-15.
- Ryachi K, Zeroual A, Khamliche L, et al. Understanding the regioselectivity and reactivity of some ethylene compounds using Parr functions. J Nat Prod Plant Resour. 2015;5:18-22.
- Zeroual A, Zoubir M, Hammal R, et al. Understanding the regioselectivity and reactivity of Friedel?Crafts benzoylation using Parr functions.Mor J Chem. 2015;3: 356-60.
- Zeroual A, Benharref A, El Hajbi A. Theoretical study of stereoselectivity of the [1+2] cycloaddition reaction between (1S,3R,8S)-2,2-dichloro-3,7,7,10-tetramethyltricyclo[6,4,0,01.3]dodec-9-ene and dibromocarbene using density functional theory (DFT) B3LYP/6-31G*(d). J Molecular Modelin.2015;21:1610-2940.
- Zeroual A, Hammal R, Ryachi K, et al. Understanding the regioselectivity and Reactivity of ß-Himachaleneusing Zeroualfunction as a new regioselectivity descriptor. Int J Innovation Appl Studies.2014;8:750-55.
- Zeroual A, El Idrissi M, Benharref A, et al. Theoretical study of regioselectivity and stereoselectivity of condensation of ß-himachalene with dichlorocarbene using density functional theory (DFT). Int J InnovAppl Stud.2014;5:120-30.
- El Idrissi M, Zeroual A, Benharref A, et al. Determination of certain thermodynamic and geometric values and condensation mechanism of ß-himachalene and dibromocarbene using density functional theory (DFT)? Phys Chem News.2013;69:89-95.