Research Article - Journal of RNA and Genomics (2017) Volume 13, Issue 1
Identification of Micro-RNA Seed Sequences and Other Possible Conserved Motifs: An Information Theoretic Approach
Md Izhar Ashraf* and A. Anny Leema
Department of Computer Applications, B. S. Abdur Rahman University, Vandalur, Chennai, India
Received date: 29 May 2017; Revised date: 07 July 2017; Accepted date: 10 July 2017; Published date: 17 July 2017
© Copyright The Author(s). First Published by Allied Academies. This is an open access article, published under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0). This license permits non-commercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details
Abstract
Micro-RNA (miRNA) is a small non-coding RNA molecule (22 nucleotides) found in plants, animals and some viruses, which regulates the expression of protein coding genes (target gene) by degrading or repressing the translation of its mRNA. For the regulation of target gene, in some cases micro RNA binds fully with mRNA, whereas in other cases it binds only through a specific conserved region in it, called the seed. These seeds having very specific location and size comprise of highly conserved nucleotide sequences. In this paper we have used a unsupervised computational technique to find out those seeds without using the biology of micro-RNA. Our method successfully managed to find 252 known seeds and also suggested a set of probable candidate for other conserved sequence, which can be used as a template for experiment.
Keywords
Sequence, Segmentation, Micro-RNA, Point wise mutual information
Introduction
The Human Genome Project has revolutionized our perspectives towards understanding biological processes. The subsequent progress in computational capabilities has given new horizons to the biologists (Ideker et al, 2001). Where initially we could only study one gene or molecule at a time, we now have come all the way up to whole genome level (DNA and RNA). We are now in a better position to understand the concept of central dogma and study it in all its intricate details, as a result of which we can envisage an era of personalized medicine (Hamburg and Collins, 2010). Deoxyribonucleic acid (DNA) is an essential part of known form of life, which carries the ‘genetic information’ and it conveys through a series of steps, called the central dogma. Specific portions of DNA, called genes, are transcribed into mRNAs which in turn convert into functional proteins that ultimately carry out the respective traits of life processes. Human DNA has around 20-25 thousand protein coding genes (International Human Genome Sequencing Consortium, 2004), but not all of them must be active simultaneously at any given time. There is an intricate system of mechanisms dedicated entirely to regulating the gene expression and those mechanisms are primarily propagated through the so called ‘non-protein coding’ genes, also called the RNA-genes, since they give rise to those RNAs that do not result into proteins. One such category of RNA is called the micro-RNA (Lu et al, 2005). These 22 (approximately) base pair long single stranded microRNAs have their respective target genes which they regulate by binding with the concerned mRNAs, completely or partially.
It has been suggested (Felekkis et al, 2010) that a single micro-RNA can regulate up to 100 different genes in humans. Approximately 30% of all the genes are regulated only through micro-RNA (Lewis et al, 2005), which makes them a center of wide spread attention over a large section of scientific community. The first reporting of miRNA was done by Victor Ambros’s lab in 1993 (Lee et al, 1993). Over the last couple of decades there has been a large body of work on micro-RNA target genes and their regulatory mechanisms.
The general mechanism of biogenesis of micro-RNA is well understood (Bartel, 2004; MacFarlane and Murphy, 2010). It originates at the nucleus from its primary transcript, called ‘primary micro-RNA’ which is later processed by an enzyme, called DROSHA and converted into a 70-100 base pair long structure called precursor micro-RNA. The precursor micro- RNA molecule then migrates from the nucleus to cytoplasm, where it is diced by another enzyme called DICER and eventually becomes a mature micro-RNA. Furthermore, in order to regulate the target genes, there exists a very specific region within the mature micro-RNA that must bind with the target mRNAs, either completely or in some cases partially. This binding region within the micro-RNA is known as seed region, which normally lies between 1st to 8th positions of micro- RNA from the 5’ end (Lewis et al, 2003). The seed regions in all the miRNAs are highly specific and their sequences are known to be con-served (Bartel, 2009). The exact position and length of miRNA is debatable (6-8 mers) but in higher mammals, consensus with 2nd to 8th position (7-mers) from the 5’ end (Lewis et al, 2005; MacFarlane and Murphy, 2010; Marin et al, 2013).
In spite of the extensive studies on miRNA, which led to obtaining a large number of seed region sequences, we are still far away from having a complete set of identified seed sequences of all the existing miRNAs in humans. In this paper, we have suggested a distinct information theoretic approach to identify specific motifs within the known miRNA sequences that might correspond to the identified seed sequences. Further on, our method gives us many more significant sub-sequences that might pro-vide us glimpses onto large number of yet undiscovered seed sequences. Our results show that a significantly large number of seed region sequences can actually be identified as motifs based on purely information theoretic segmentation of the known miRNA sequences. We also determine a large number of unidentified motifs, which might serve as possible candidates for the unknown seed region sequences.
Materials and Methods
For our analysis we have used two kinds of data sets, one is mature micro-RNA data of Humans and the other one is micro-RNA seed data of same. Mature micro-RNA data has been collected from miRBase (Griffiths-Jones et al, 2008), which is a micro-RNA sequence and annotation database. It is managed by Griffiths-Jones lab at the Faculty of Life Sciences, University of Manchester. In the database we found 2588 different mature micro-RNA sequence of humans whose average length is 22 base pair long with minimum 16 and maximum 28 base pair (date of access: June 23rd, 2016). The other data (Human micro-RNA seed) has been collected from TargetScan (Agarwal et al, 2015) (date of access: July 7th, 2016), which is a web server that predicts biological targets of micro-RNAs by searching for the presence of sites that match the seed region of each miRNA. This server is regularly updated and improved by the laboratory of David Bartel and the Bioinformatics & Research Computing Group of Whitehead Institute. Here we acquired 2061 human micro-RNA seeds, which we are using to compare with our predicated conserved motif.
Since all the known seed sequences are 7 nucleotide long, we were interested only in obtaining conserved motifs that are specifically 7-mers. We segmented the mature micro-RNA sequences into significant sub sequences based on the mutual information. Mutual information, which is a measure of association (Martin and Jurafsky, 2015), was developed by Church and Hanks (Church and Hanks, 1989). The point wise mutual in-formation (PMI) tells us how often two events x and y occur simultaneously, with respect to what is expected when they are independent events. In contrast of mutual information(MI), PMI refers to single events, whereas MI refers to the average of all events. In other words it is a quantitative measure of joint occurrence probability of two events divided by multiple of independent probabilities of their occurrence. It is computed as:
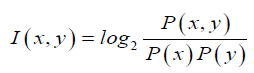
For segmentation of a given miRNA sequence, first we calculated the point wise mutual information of each consecutive pair of nucleotides within that miRNA sequence by using above formula, while considering the all pair statistics of the entire miRNA database. Then we replace the high PMI-scoring pair of empirical miRNA sequence with a new meta-sign Xi (where i is number of replacement) and the resulting sequence length will be one less than the original sequence length. We repeat the above procedure until the full sequence gets converted into a single new meta-sign. If sequence length is l then number of replacements will be l 1. Then we unfold the meta-sign into a full binary tree, as represented by Figure 1, where each node (Xi) is a result of each meta-sign replacement of high PMI-scoring pair. Here leaf nodes indicate the empirical miRNA sequence whereas other nodes than leaf nodes represents the different sub-sequence of different length. But as mentioned above, in our study we are only focused on the nodes with sub-sequences of length 7 (in presented Figure 1 node number X11).

Figure 1. Segmentation tree of a micro-RNA sequence of human genome. Segmentation has been done based on the mutual information (measure of association, detail described in method section II) of adjacent nucleotide pairs of micro-RNA sequence. Each node (meta-sign xi 18) represents the sub sequence(motif) while all have also mentioned with its significance level, p-value.
Results
Our study segments all mature micro-RNA sequences into several significant sub-sequences based on their association (point wise mutual information). These sub-sequences can vary in length. In accordance with the known length of seed regions of miRNA, we only consider the sub-sequences of length 7. Figure 1 shows the tree node number X11 representing a sub sequence of length 7. For the full database (described in material and methods section) we got total 1126 7 mers, where many of them appear multiple times, thus the total unique number of 7 mers is 754. Out of all these 754 7 mers, a significant 252 match fully with the known micro-RNA seeds, which is almost one third of total number of significant 7 mers (252 out of 754). This has been schematically represented in Figure 2 by a Venn diagram. Figure 3 represents the number of embedded known seed, if we consider n mers 7 or more. The maximum length of n mers we have considered is 16 because; the minimum length of mature micro-RNA obtained from miRBase for our study is 16 nucleotide long.

Figure 2. Venn diagram representing overlap between set of known seed region sequences and the set of significant 7-mers identified using segmentation method. This figure shows that 1/3rd of identified 7-mers are present in the known seed list.

Figure 3. Overlap between seed sequences and significant segments of varying length The gure shows number of known seed sequences which were found embedded within the significant segments of different lengths obtained through our method. ,where x axis represent the number of nucleotide of length n, which varies 7-16 whereas y axis rep-resents the number of known 7 mers found in the significant segment found by our method. Though our study mainly focused on segments of length 7, since they can directly compare with the known seed sequence, which are of length 7.
Conclusions
In our study we found that almost one third of the significant 7-mers are found in the known set of seed sequences (252 out of 754). It is a good validation to the significant sub-sequences, which are conserved over all the miRNA sequences, being good candidates to be situated in the seed regions. This makes even the remaining sequences (754-252) equally interesting since we can reasonably expect those remaining sequences also to be of equivalent importance given that they too are highly con-served over the miRNA sequences. Hence, many of those sequences might actually be occurring in the seed regions of unidentified miRNAs. The novelty of this work is even though being highly conserved is not a definitive evidence for a sequence to be a seed sequence, nevertheless using this method we can narrow down the number of possible candidates for the yet to be discovered seed sequences and we managed to get it without using any positional information whereas earlier methods are mainly based on position. Our method gives a set of significant sub-sequence, among which one third are seed sequence, furthermore, even if some of the significant sub-sequences do not actually occur in the seed regions, the high degree of conservation in these sequences strongly suggests some form of latent functional importance to them, which is yet to be found out through experiment.
Acknowledgments
We thank Anand Pathak, Janaki Raghavan and Soumya Eswaran for their helpful discussions, suggestions and comments on manuscript.
References
- Agarwal V, Bell GW, Nam JW, et al. 2015. Predicting effective microRNA target sites in mammalian mRNAs. Elife, 4, e05005.
- Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 116, 281-297.
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell, 136, 215-233.
- Church KW and Hanks P. 1989. Word association norms, mutual information, and lexicography, Proceedings of the 27th Annual Meeting of the Association for Computational Linguistics.
- Felekkis K, Touvana E, Stefanou CH, et al. 2010. microRNAs: a newly described class of encoded molecules that play a role in health and disease. Hippokratia, 14, 236.
- Griffiths-Jones S, Saini HK, van Dongen S, et al. 2008. miRBase: tools for microRNA genomics. NAR 36. Database Issue), D154–D158.
- Hamburg MA and Collins FS. 2010. The path to personalized medicine. N Engl J Med, 363, 301-304.
- Ideker T, Galitski T and Hood L. 2001. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet, 2, 343-372.
- International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature, 431, 931-945.
- Lee RC, Feinbaum RL and Ambros V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75, 843-854.
- Lewis BP, Burge CB and Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell, 120, 15-20.
- Lewis BP, Shih IH, Jones-Rhoades MW, et al. 2003. Prediction of mammalian microRNA targets. Cell, 115, 787-798.
- Lu J, Getz G, Miska EA, et al. 2005. MicroRNA expression profiles classify human cancers. Nature, 435, 834-838.
- MacFarlane LA and R Murphy P. 2010. MicroRNA: biogenesis, function and role in cancer. Curr Genomics, 11, 537-561.
- Marín RM, Šulc M and Vaní?ek J. 2013. Searching the coding region for microRNA targets. RNA, 19, 467-474.
- Martin, JH and Jurafsky D. 2015. Speech and language processing. International Edition, 710.