Research Article - Biomedical Research (2016) Computational Life Sciences and Smarter Technological Advancement
Analysis of multilayer perceptron machine learning approach in classifying protein secondary structures
Leo Dencelin X1* and Ramkumar T21Department of Computer Science and Engineering, Sathyabama University, Chennai, India
2School of Information Technology and Engineering, VIT University, Vellore, India
- *Corresponding Author:
- Leo Dencelin X
Department of Computer Science and Engineering
Sathyabama University
India
Accepted date: August 29, 2016
Abstract
Protein secondary structure prediction is an important problem in bioinformatics and transforming biomedical big data into valuable knowledge is also a quite interesting and challenging task. Various machine learning algorithms have been widely applied in bioinformatics to extract knowledge from protein data. In recent years, multilayer perceptron, emerging on the basis of artificial neural networks, is making major advances in many domains. In this paper, we have focused on analysing the machine learning based Multilayer Perceptron (MLP) algorithms in protein secondary structure prediction using different set of input features and network parameters in distributed computing environment. Overall, the MLP analysis results in classifying protein secondary structures are encouraging, the accuracy and performance are overwhelming by passing various input features and it outperforms when it will be implemented in various distributed environment. The experimental result shows that the multilayer perceptron machine learning algorithm models outperforms the other machine learning approaches.
Keywords
Machine learning, Multilayer perceptron, Back propagation, Protein secondary structure prediction, Distributed framework, Apache spark, Position-specific scoring matrix, Confusion matrix.
Introduction
Proteins are important for living organisms. For example, our body fabric is made of protein molecules. They serve as hormones, receptors, storage, defence, enzymes and as transporters of particles in our bodies. Proteins are made of simple building blocks called amino acids. There are 20 different amino acids that can occur in proteins. Their names are abbreviated in a three letter code or a one letter code and vary significantly. These amino acids that make up proteins can be grouped according to many criteria, including hydrophobicity, size, aromaticity, or charge. There are four different structure types of proteins. Primary structure refers to the amino acid sequence of a protein. It provides the foundation of all the other types of structures. Secondary structure refers to the arrangement of connections within the amino acid groups to form local structures. Α-helix, β-strand are some examples of structures that form the local structure. Tertiary structure is the three dimensional folding of secondary structures of a polypeptide chain. Quaternary structure is formed from interactions of several independent polypeptide chains. A typical protein contains about 32% alpha helices, 21% beta sheets and 47% loops or non-regular structures [1]. Theoretically, it is not possible to predict 100% accurate protein structure because of the fact that there are 20 different amino acids and thus no. of ways to generate similar structure in proteins by different amino acids is much more. The function of a specific protein is mostly known from its molecular structure. Tertiary Structure Prediction (TSP) can determine the structure of the viral proteins which leads to the design of drugs for specific viruses. TSP provides structure function relationship. It means that a particular protein structure is responsible for a particular function. So by changing the structure of the proteins or by synthesizing new proteins, functions could be added or removed or desired functions could be obtained [2].
In our work, an artificial neural network based machine learning approach has been proposed in which MLP’s are trained to make them capable of recognizing the primary sequences and the Dictionary of Protein Secondary Structure (DSSP) codes of the protein structures and their association with the secondary structure is derived (Figure 1). Ann’s function as a two level classifier for the proposed work. Some of the previous works done are [3]. Although various PSSP has been widely adopted in industry, the research on PSSP is still at an early stage. Many existing issues have not been fully addressed, while new challenges keep emerging in PSSP research. Data mining in bioinformatics is hampered by many facets of biological databases, including their size, number, diversity and the lack of a standard ontology to aid the querying of them as well as the heterogeneous data of the quality and provenance information they contain. Another problem is the range of levels the domains of expertise present amongst potential users, so it can be difficult for the database curators to provide access mechanism appropriate to all. The integration of biological databases is also a problem. Data mining using machine learning and proteomics are fast growing research area today. It is important to examine what are the important research issues in proteomics and develop new data mining methods for scalable and effective analysis.

Figure 1: Primary and secondary structures.
Structure Prediction Concepts and Techniques
Amino acids
Each amino acid is identified by its side chain which determines the properties of amino acid. Amino acids are separated into four groups non-polar, polar, basic, and acidic. Polar and non-polar are again categorized under hydrophobic (attracted towards water) and hydrophilic (repelled by water). The combination of the properties that allow a specific protein to form into a certain structure is not completely known. There are many inherent properties that amino acids have that are involved in determining the structure of a protein. One of the most important distinguishing factors of amino acids is their different tails which are also called the R groups [4]. Other factors play key roles in determining the final structure of a protein, these include: the energy level of the structure which needs to be low and stable and links between amino acids. A protein does not exhibit a full biological activity until it folds into a three-dimensional structure [5]. The goal is to determine the shape (fold) that a given amino acid sequence will adopt. Due to the size, shape and charge of its side chain, each amino acid may “fit” better in one type of secondary structure than other. The list of amino acids and symbols denoted is given in Table 1.
| S. no | Amino acids | Symbols | |
|---|---|---|---|
| 1 | Alanine | Ala | A |
| 2 | Cysteine | Cys | C |
| 3 | Aspartic acid | Asp | D |
| 4 | Glutamic acid | Glu | E |
| 5 | Phenylalanine | Phe | F |
| 6 | Glycine | Gly | G |
| 7 | Histidine | His | H |
| 8 | Isoleucine | Ile | I |
| 9 | Lysine | Lys | K |
| 10 | Leucine | Leu | L |
| 11 | Methionine | Met | M |
| 12 | Asparagine | Asn | N |
| 13 | Proline | Pro | P |
| 14 | Glutamine | Gln | Q |
| 15 | Arginine | Arg | R |
| 16 | Serine | Ser | S |
| 17 | Threonine | Thr | T |
| 18 | Valine | Val | V |
| 19 | Tryptophan | Trp | W |
| 20 | Tyrosine | Tyr | Y |
Table 1: Names and symbols of 20 amino acids.
Several studies have shown that a stronger correlation exists between structure conservation and function, i.e. structure implies function and a higher correlation exists between sequences and structure (Sequence->structure->function). On other words, proteins with similar sequences and structures have similar functions. Moreover, similar sequences in proteins imply that they also have similar structures.
Every protein has a unique linear sequence of amino acids, also called a polypeptide. This amino acid sequence contains information that guides the protein to fold up into a unique shape. To be able to perform their biological function, proteins fold into one or more specific spatial conformations. To understand the functions of proteins at a molecular level, it is often necessary to determine their 3D structure. The tertiary structure is the 3D fold of the protein molecule comprising of secondary structure elements: alpha (α) helices, beta (β) sheets and loops. The Dictionary of Protein Secondary Structure (DSSP) is commonly used to describe the protein secondary structure with single letter codes.
Ann based machine learning techniques
Machine Learning refers to the techniques involved in dealing with vast data in the most intelligent fashion (by developing algorithms) to derive actionable insights. This is of immense importance in bioinformatics and in particular, supervised machine learning has been used to great effect in numerous bioinformatics prediction methods [6,7]. Machine learning methods that can automatically extract knowledge from the Protein Data Bank (PDB) are an important class of tools and have been widely used in all aspects of protein structure prediction. Various studies show that, the Artificial Neural Network (ANN) is not only the dominant league leader in 2011 but has been in this position since at least the 1970’s. Based on the research requirements, the machine learning techniques were used in combination with each other [8].
Artificial Neural Network (ANN) is made up of interconnecting artificial neurons. The general function of a neural network is to produce an output pattern when given a particular input pattern, and is loosely related to the way the brain operates. Learning these mappings is done in conceptually the same way as the brain. Several types of neural networks exist but the most common one used has been the multi-layer perceptron. Another ANN used in the work is Radial Basis Function (RBF) which is faster compared to MLP. The RBF uses a Bayesian decision making to process applied patterns [9]. It has two hidden layers of which the first one provides a class distribution probability while the second one provides a decision depending upon the closeness the applied patterns shall have using a Gaussian spread function. Recently deep learning techniques are showcasing more accuracy in proteomics field [10,11]. Multilayer Perceptron Artificial Neural Networks (MLP-ANNs) are computational models which are popular means to model complex relationships between inputs and outputs and to find patterns. In supervised learning the MLP class of neural networks requires a set of training samples which are used to infer a classifier to predict a correct output value. To avoid over fitting and to assess the robustness of ANN class assignment , the general strategy of ANN analysis routinely included procedure of training, internal validations and testing , ideally under blinded conditions [12]. ANN performance can be optimized by modifying the way of pre-processing, adding or eliminating the spectral features or changing the networks architecture [13]. The purpose of neural network training is to minimize the output errors on a particular set of training data by adjusting the network weights (w). We define a cost function E (w) that measures how far the current network’s output is from the desired one. Partial derivatives of the cost function ∂ E (w)/∂ w tell us which direction we need to move in weight space to reduce the error [14]. The learning rate η specifies the step sizes we take in weight space for each iteration of the weight update equation. We keep stepping through weight space until the errors are ‘small enough’. Various weight update rules Generalized Delta Rule (GDR or Back propagation), gradient descent rule are being used to minimise the error rates (Figures 2 and 3).

Figure 2: A Multilayer perceptron with four hidden layers and three output layer.

Figure 3: Proposed architecture of multilayer perceptron classifier.
Methodology Used
This Artificial neural network based MLP classification model aims to classify the three types of secondary structures α-helix, β-sheet and coils from the PDB dataset. The input features considered for this model are 1) PSSM profile generated using PSI-BLAST approach, 2) amino acid sequence and 3) physiochemical properties. The protein dataset was a collection of different proteins from PDB [15] and the accuracy measures sensitivity, precision, F1 score were derived from the confusion matrix.
Encoding and feature engineering
Dictionary of Protein Secondary Structure (DSSP) is a database of secondary structure assignments for all protein entries in the Protein Data Bank (PDB). In there are 8 secondary structures of protein available. These structure include H (alpha-helix), G (3-helix or 310-helix), I (5-helix or p-helix), B (residue in isolated beta-bridges), E (extended sheet), T (hydrogen bond turn), S (bend) and “-“(any other structure). Typically these 8 structures are reduced to three main classes which are of more practice and use [16]. There are different rules applied in the literature to perform such mapping. Table 2 shows existing rules to convert 8 structures to 3 states and these rules was applied to the training, testing and validation datasets.
| Rule# | Rules |
|---|---|
| Rule 1 | {H, G} to {H} { E, B} to {E} {All other states} to {C} |
| Rule 2 | {H} to {H} {E} to {E} {All other states} to {C} |
| Rule 3 | {H, G, I} to {H} {E} to {E} {All other states} to {C} |
| Rule 4 | {H, G} to {H} {E} to {E} {All other states} to {C} |
| Rule 5 | {H, G, I} to {H} {E, B} to {E} {All other states} to {C} |
| Rule 6 | {H,G,I} to {H} {E,B} to {E} {All other states} to {L} |
Table 2: Conversion rules followed.
| Parameters | Values/techniques used to improve MLP performance |
|---|---|
| Activation function | Hyperbolic tangent function |
| No. of hidden neurons | 150 |
| No. of hidden layers | 50 |
| Maximum no. of epochs | 1651 |
| Error function | CE cost function |
| Learning rate | 0.1 |
| Window size | 10 |
| Input normalization | Input variables are modified to be uncorrelated, same variance and mean value to zero/small |
| Initial weights | Hidden-to-Output (HO) weights is larger than Input-to-Hidden (IH) weights |
| Weight decay | added decay term = w (n+1) = (1-ε)*w (n) |
| Weight updates | Batch training |
Table 3: Network parameters MLP parameters used in our analysis.
Feature transformation: PSSM profiles
A Position Weight Matrix (PWM), also known as a Position- Specific Weight Matrix (PSWM) or Position-Specific Scoring Matrix (PSSM), is a commonly used representation of motifs (patterns) in biological sequences. PWMs are often derived from a set of aligned sequences that are thought to be functionally related and have become an important part of many software tools for computational motif discovery [17]. Since PSSM profiles are involved with biological evolution, we consider them as features in our work. This matrix is based on the frequencies of each residue in a specific position of a multiple alignment and has 20 × L elements, where L is the length of a query sequence. Protr package in R statistical programming is used to extract the PSSM feature from the sequence. PSSM scores are generally shown as positive or negative integers. Positive scores indicate that the given amino acid substitution occurs more frequently in the alignment than expected by chance, while negative scores indicate that the substitution occurs less frequently than expected. Two methods namely 1) simple piecewise function with a linear distribution, 2) logistic function were used by most of the researchers for scaling PSSM values to the range (0, 1). We have used the first one in this analysis to scale the PSSM values as the simple piecewise function ignores scoring differences at the extremities and linearly scales the intermediate scores.
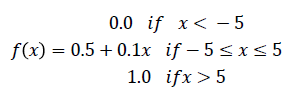
Feature selection: physio-chemical properties
The Physical and chemical properties of protein aids to determine the protein structure, it has been used rigorously to distinguish native or native like structure from other predicted structures. Here we have explained the features and properties that were mainly used in structure prediction methodologies. 1) Net charge: One of the physical properties of amino acids is their charges. Five of the amino acids are charged amino acids: R, D, E, H, and K. Residues which have similar electric charge repel each other and it interrupts the hydrogen bonds in the main chain of amino acids. It prevents the formation of α-helix. In addition, continues β-sheet formation is not possible when the residues have similar charges. This physical property of amino acids helps to predict secondary structure of proteins [18]. 2) Hydrophobicity: Few amino acids do not like to reside in an aqueous environment and they called hydrophobic amino acids. They are generally seen buried within the hydrophobic core of protein since for protein folding, polar residues prefer to stay outside of protein in order to prevent non polar residues from exposing to polar solvent. Hydrophobic protein can be used as one of the parameter to predict the secondary structure of proteins. In α-helix, generally hydrophobic segments are followed by hydrophilic segment. Unlike α-helix, β-sheet structure is affected by the environment due to its structural characteristics so it is not a case in β-sheets. 3) Side chain mass: Although the basic structure is the same for 20 amino acids, the size of the side chain R group still influences structure folding. Side chains of amino acids are the structural elements which make amino acids different. Unique R groups of the amino acids, influencing the conformation of protein secondary structure and they can give a clue to predict the secondary structural element depends on their existence in certain position. The side chain R group form in the outside of the main chain of α- helix structure but when large R groups distributed continuously, they can make α-helix structure unstable.
Multilayer perceptron based classification model
A Multilayer Perceptron (MLP) is a feed-forward artificial neural network model that maps sets of input data onto a set of appropriate output. An MLP consists of multiple layers of nodes in a directed graph, with each layer fully connected to the next one. Except for the input nodes, each node is a neuron (or processing element) with a nonlinear activation function [19]. MLP utilizes back propagation for training the network. This class of networks consists of multiple layers of computational units, usually interconnected in a feed-forward way. In many applications the units of these networks apply a sigmoid function as an activation function.
We have considered the below termination conditions in MLP classifier:
• Fixed number of iterations.
• Error falls below some threshold.
• Error on a separate validation set falls below some threshold.
Learning rule: Back propagation algorithm
A learning rule is applied in order to improve the value of the MLP weights over a training set T according to a given criterion function. Back propagation algorithm (Figures 4 and 5) is a supervised learning method which is applied to train the fully connected feed forward network and consists of two phases: 1) forward phase where the activations are propagated from the input to the output layer. This phase propagate inputs by adding all the weighted inputs and then computing outputs using sigmoid threshold, and 2) backward phase, where the error between the observed actual and the requested nominal value in the output layer is propagated backwards in order to modify the weights and bias values [20]. This phase propagates the errors backward by apportioning them to each unit according to the amount of this error the unit is responsible for. The network weights are modified towards minimizing the square error of the network output. The weight matrices are updated in every position, respectively. The network weights are initialized with small random values within (-0.1, 0.1) interval. Training is terminated when either the relative error reduces to less than 0.1 or the training epochs reach up to 1000. At each training epoch, the samples of the training set are fed in randomly changing orders.

Figure 4: Back propagation algorithm used in this analysis to minimize the errors.

Figure 5: Line graph shows the variations with no. of hidden neurons vs. accuracy measures considered in this analysis.
Training and algorithm tuning
We have trained our MLP using 5045 different proteins from Protein Data Bank (PDB), with the total number of amino acid as 105,000. 70:30 split ratio rule was applied to fetch the data for training, validation and testing phase, which uses 70% of the amino acids for training and 30% for both testing and validation. Machine learning algorithms are driven by parameters. These parameters majorly influence the outcome of learning process. The objective of parameter tuning is to find the optimum value for each parameter to improve the accuracy of the model [21]. To tune these parameters, we must have a good understanding of these meaning and their individual impact on model. A MLP trained with back propagation will generally train faster if the activation function is anti-symmetric f (-x) =-f (x) [22]. Considering this, we have used the hyperbolic tangent function as activation function here. Our Analysis uses some techniques to normalize the input variables to achieve these conditions 1) mean value is zero or small compared to the variance 2) uncorrelated 3) same variance. This MLP analysis uses small random weights to avoid driving the neurons into saturation. The Hidden-to- Output (HO) weights were made larger than Input-to-Hidden (IH) weights since they carry the back propagated error. If the initial HO weights are very small, the weight changes at the IH layer will be initially very small, slowing the training procedure. The number of hidden units determines the degrees of freedom or expressive power of the model and we have assigned 5 hidden neurons within each hidden layers [23]. To prevent the weights from growing too large (a sign of overtraining) we have added a decay term of the form w (n+1) = (1- ε)*w (n). By doing so, weights that are not needed eventually decay to zero, whereas necessary weights are continuously updated by back propagation. The two basic approaches for updating the weights during training are 1) On-line training: weights are updated after presentation of each example 2) Batch training: weights are updated after presentation of all the examples (we store the Δ w for each example, and add them up the weight after all the examples have been presented). This analysis uses batch training as it uses the true steepest descent direction. Below is the summary of parameters tuned to attain the accuracy of the constructed multilayer perceptron classifier.
Accuracy evaluation
The evaluation measures applied in our analysis uses the confusion matrix parameters [24] namely TP, TN, FP and FN described below. Caret package in R programming is used to measure the accuracies.
TP (True positive): number of correctly predicted residues for each class.
TN (True Negative): number of correctly predicted residues not belonging to each class.
FP (False positive): number of incorrectly predicted residues to belong to each class.
FN (False Negative): number of predicted residues not to belong to each class.
The below terminology and derivations from confusion matrix are used to measure the accuracy of our analysis (Table 4).
| Actual | Predicted | |||
|---|---|---|---|---|
| Helix (H) | Sheet (E) | Coil (C) | ||
| Helix (H) | 75.80% | 10.67% | 8.77% | |
| Sheet (E) | 15.57% | 78.43% | 3.68% | |
| Coil (C) | 14.76% | 2.97% | 82.74% | |
Table 4: Confusion matrix proportion of predicted secondary structures.
Sensitivity: Sensitivity, also called the true positive rate measures the proportion of positives that are correctly identified as such.
Sensitivity (TPR) =TP/ (TP+FN)
Precision: Precision is the number of true positives divided by the number of true positives and false positives. Precision can be thought of as a measure of classifiers exactness. It is also called the Positive Predictive Value (PPV).
Precision (PPV) = TP/ (TP+FP)
F1 score: F1 score is also called the F score or the F measure and conveys the balance between the precision and the recall.
F1 Score = 2 TP/ (2TP+FP+FN)
Prediction performance for individual secondary structure states are shown in Table 3. The sensitivity, precision, and F1 score are calculated across amino acid residues of all proteins in CB513 dataset are shown below. The secondary structure helix has the highest value of 0.912, 0.845, 0.452 for sensitivity, precision and F1 score respectively and has shown a high accuracy on CB513 dataset (Table 5).
| Secondary structure | Sensitivity (TPR) | Precision (PPV) | F1 score (Harmonic mean of Sensitivity and Precision) |
|---|---|---|---|
| H-helix | 0.912 | 0.845 | 0.452 |
| E-sheet | 0.862 | 0.651 | 0.326 |
| C-coil | 0.613 | 0.543 | 0.141 |
Table 5: Classification accuracies sensitivity, precision and F1 Score to detect the positive rate.
| Features and parameters used | Accuracy (%) |
|---|---|
| With PSSM | 68.20% |
| With PSSM and physiochemical features | 69.67% |
| With input normalization | 65.55% |
| Without input normalization | 60.64% |
| With more than three hidden layers | 68.60% |
| MLP with weight decay term added | 67.56% |
| MLP with batch training | 67.20% |
| MLP with online-training | 63.32% |
Table 6: An ablation study to identify the impact of features and parameters of our analysis.
We have performed an ablation study to discover the key features and parameters involved in our MLP analysis. For this, we have removed and replaced the various components used in this analysis and tested the model performance by having position specific scoring matrix alone as input, PSSM and physio-chemical properties, normalized the input features to be uncorrelated and modified the mean value to zero and small, adding weight decay term and batch and online training for better error reduction. From the results on CB513 dataset, PSSM along with physiochemical properties has achieved 69.67% of accuracy and the results are convincing for normalized input. Also the batch training mode has achieved 67.20% of accuracy when compared to online-training mode which was only 63.32%.
Distributed framework
A distributed system is a setup in which several independent computers (computing nodes) participate in solving the problem of processing a large volume of and variety of structured/semi-structured/unstructured data [25]. Currently, big data is one of the hottest topics in computer science, due to the rapid increase of the amount of data we, as a society, produce and store. The driving force behind this increase is a dramatic drop in the costs of collecting and storing this data. This decrease in costs is not only observed in social data (behaviour) and the internet of things (sensors) but also in the area of proteomics. This means that the amount of proteomics data is growing faster than the capacity to do computations on it. At the same time we would like to have our results faster, for example to start a treatment sooner rather than later. To be able to obtain maximum value from this data we need a system that can scale as the data grows [26-28]. The current best implementation of the well-known map-reduce-paradigm is apache spark, which optimizes the computations by making better use of memory than Hadoop, the previous De facto open source choice for doing map-reduce [29]. Spark is a general purpose, open source cluster computing framework and can be used for any kind of map-reduce computation. It was built on top of Hadoop-Map reduce and it extends the map reduce model to efficiently use more types of computations which includes interactive queries and stream processing. At the core of spark is the notion of a Resilient Distributed Dataset (RDD), which is an immutable collection of objects that is partitioned and distributed across multiple physical nodes of a Yet Another Resource Negotiator (YARN) cluster and that can be operated in parallel. Spark is an ideal platform for organizing large proteomics analysis pipelines and workflows [30].
We have used apache spark ML lib API’s for implementation of Multilayer Perceptron Classifier (MLPC) and are available in classification and regression package of ML lib. MLPC employs back propagation for learning the model. Reasons to choose spark are, 1) Spark uses the concept of RDD which allows us to store data on memory and persist it as per the requirements [31]. This allows a massive increase in batch processing job performance 2) Spark also allows us to cache the data in memory, which is beneficial in case of iterative algorithms such as those used in machine learning. 3) Spark allows us to perform stream processing with large input data and deal with only a chunk of data on the fly [32]. This can also be used for online machine learning, and is highly appropriate for use cases with a requirement for real time analysis which happens to be an almost ubiquitous requirement in the industry.
Our aim is also to improve the performance of the approach along with accuracy on predicting the protein secondary structures. Spark being a distributed processing system helped us on enhancing the performance.
Conclusion
In this paper we present a neural network based MLP classifier to predict the secondary structure of protein. We have analysed the classifier accuracy with varied set of parameters by changing the input dataset features, perceptron parameters and generated the prediction results. The MLP parameters are modified and the prediction results were compared for accuracy. MLP configured with more number of hidden layers, normalized input features, adding a weight decay term have attained a good accuracy when compared to other amino acid features and network parameters. This work was also implemented in distributed environment for their scalable, efficient computing capacity, bandwidth, storage, security, and reliability and fault-tolerant/robust features. The performance of the classifier is increased while implemented in distributed framework Spark. As a conclusion, MLP together with spark will be more popular in the future as both accuracy and speed considerations are likely to remain important as proteomic projects continue to generate great challenges/opportunities in this area.
References
- Qu W, Sui H, Yang B, Qian W. Improving protein secondary structure prediction using a multi-modal BP method. Comput Biol Med 2011; 41: 946-959.
- Zhang H. Protein tertiary structures: prediction from amino acid sequences. Encyclop Life Sci 2002.
- Gutachter, Martin V, Walaa F. Approaches to protein structures. Freie Univ Berlin 2011.
- Luo RY, Feng ZP, Liu JK. Prediction of protein structural class by amino acid and polypeptide composition. Eur J Biochem 2002; 269: 4219-4225.
- Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 2001; 43: 246-255.
- Chou KC. A novel approach to predicting protein structural classes in a (20-1)-d amino acid composition space. Proteins 1995; 21: 319-344.
- Chou KC. A key driving force in determination of protein structural classes. Biochem Biophys Res Commun 1999; 264: 216-224.
- Rajkumar B, Duzlevski O, Xu D. Profiles and fuzzy k-nearest neighbour algorithm for protein secondary structure prediction. APBC 2005.
- Jo T, Hou J, Eickholt J, Cheng J. Improving protein fold recognition by deep learning networks. Sci Rep 2015; 5: 17573.
- Lena P, Nagata K, Baldi P. Deep spatio-temporal architectures and learning for protein structure prediction. Adv Neur Inform Proc Sys 2012; 25: 521-529.
- Bengio, Yoshua, Thibodeau L. Deep generative stochastic networks trainable by backprop. ArXiv 2013; 1306: 1091.
- Zhiyong W, Feng Z, Jian P, Jinbo X. Protein 8 class secondary structure prediction using conditional neural fields. Proteom 2011; 11: 3786-3792.
- Fakoor R, Ladhak F, Nazi A, Huber M. Using deep learning to enhance cancer diagnosis and classification. Proc Int Conf Mach Learn 2013.
- Zhou J, Troyanskaya O. Deep supervised and convolutional generative stochastic network for protein secondary structure prediction. Proc Int Conf Mach Learn 2014.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN. The Protein Data Bank. Nucleic Acids Res 2000; 28: 235-242.
- David JT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999; 292: 195-202.
- Li D, Li T, Cong P, Xiong W, Sun J. A novel structural position specific scoring matrix for the prediction of protein secondary structures. Bioinform 2012; 28: 32-39.
- Toussaint NC, Widmer C, Kohlbacher O, Ratsch G. Exploiting physico-chemical properties in string kernels. BMC Bioinformatics 2010; 11 Suppl 8: S7.
- Spencer M, Eickholt J, Cheng J. A deep learning network approach to ab initio protein secondary structure prediction. IEEE Transactions on Comp Biol Bioinformatics 2014.
- Bordoloi H, Sarma KK. Protein structure prediction using multiple artificial neural network classifiers. Comp Tech Vision Sci Computational Intellig 2012; 395: 137-146.
- Deka A, Bordoloi H, Sarma KK. ANN-aided tertiary protein structure prediction using certain coding techniques and known secondary structures. Proc Int Conf Electr Commun Eng 2012.
- Deka A, Sarma KK. Soft computational framework for tertiary protein structure prediction. Int J Electr Sig Sys 2012; 48: 33-37.
- Deka A, Sarma KK. Tertiary protein structure prediction using artificial neural network as a two-level classifier. Proc Int Conf Comp Commun Technol 2012.
- Bengio Y. Practical recommendations for gradient-based training of deep architectures. Neur Netw Trade Spr 2012; 437-478.
- Marek S, Wiewiorka, Messina A, Pacholewska A, Maffioletti S, Gawrysiak P, Michal Sparkseq-fast, scalable, cloud-ready tool for the interactive genomic data analysis with nucleotide precision. Bioinformatics 2014; 30: 2652-2653.
- Apache Spark documentation 2014.
- Machine learning with spark-spark summit 2013.
- Davidson A, Andrew OR. Optimizing shuffle performance in spark. Technical Rep Berkleey 2015.
- Spark job flow-data bricks. Data bricks 2016.
- Davison A. A deeper understanding of spark internals. Spark internals Summit 2014.
- Zaharia M, Chowdhury M, Das T, Dave A, Justin MA, McCauley M, Michael JF, Shenker S. Resilient distributed datasets: a fault-tolerant abstraction for in-memory cluster computing. NSDI 2012.
- Zaharia M, Chowdhury M, Das T, Dave A, Justin MA, McCauley M, Michael JF, Shenker S, Stoica I. Spark-cluster computing with working sets. Hot Cloud 2010.