Research Article - Biomedical Research (2017) Volume 28, Issue 3
Discriminative deep belief networks for microarray based cancer classification
Esra Mahsereci Karabulut1 and Turgay Ibrikci2*1Technical Sciences Vocational School, Gaziantep University, 27310 Gaziantep, Turkey
2Department of Electrical-Electronics Engineering, Cukurova University, 01330 Adana, Turkey
- *Corresponding Author:
- Turgay Ibrikci
Department of Electrical-Electronics Engineering
Cukurova University, Turkey
Accepted on July 07, 2016
Abstract
Accurate diagnosis of cancer is of great importance due to the global increase in new cancer cases. Cancer researches show that diagnosis by using microarray gene expression data is more effective compared to the traditional methods. This study presents an extensive evaluation of a variant of Deep Belief Networks - Discriminative Deep Belief Networks (DDBN) - in cancer data analysis. This new neural network architecture consists Restricted Boltzman Machines in each layer. The network is trained in two phases; in the first phase the network weights take their initial values by unsupervised greedy layer-wise technique, and in the second phase the values of the network weights are fine-tuned by back propagation algorithm. We included the test results of the model that is conducted over microarray gene expression data of laryngeal, bladder and colorectal cancer. High dimensionality and imbalanced class distribution are two main problems inherent in the gene expression data. To deal with them, two preprocessing steps are applied; Information Gain for selection of predictive genes, and Synthetic Minority Over-Sampling Technique for oversampling the minority class samples. All the results are compared with the corresponding results of Support Vector Machines which has previously been proved to be robust by machine learning studies. In terms of average values DDBN has outperformed SVM in all metrics with accuracy, sensitivity and specificity values of 0.933, 0.950 and 0.905, respectively.
Keywords
Cancer classification, Gene expression data, Discriminative deep belief networks, Support vector machines, Feature selection.
Introduction
Cancer is the second-leading cause of death in the United States, coming after the heart disease [1]. The name cancer refers to more than a hundred diseases characterized by out of control growth and multiplication of the cells. For the purpose of cancer diagnosis, use of microarray technology along with computer aided methods is increasing rapidly. DNA microarray technology generates features for monitoring expression of genes on genomic level for researching cancer. Diagnosis from such a microarray gene expression data is shown to be more effective compared to traditional methods [2-4]. Analysis over gene expression data for the purpose of diagnosis can be done by using additional laboratory experiments, but such experiments are costly and labor intensive. Alternatively, as a replacement to experiments for diagnosis methods from artificial intelligence can also be utilized to perform computer-based diagnosis of the gene expression data. As computer-based diagnosis has advantages over laboratory experiments such as low cost and diagnosis speed, research over computer-based methods has been gaining attention and remains essential [5].
In the literature, several machine learning techniques are evaluated for computer-based diagnosis by using gene expression profiles. The first study that belongs to Golub et al. [6] is about clustering Acute Myeloid Leukemia (AML) and Acute Lymphoblastic Leukemia (ALL) data by self-organizing maps (SOM). The subsequent studies include specific algorithms with application to specific gene expression profiles [7-9] and comparative analysis of different methods [10-13]. Support Vector Machine (SVM) is the most prominent of the methods. Its ability of handling high dimensional data, lead to successful application for classification of microarray gene expression data [14-18]. Pirooznia et al. [19] achieved a comparative study among several machine learning methods on microarray gene expression data from different cancer types. They employed methods of SVM, RBF Neural Nets, MLP Neural Nets, Bayesian Network, Decision Tree and Random Forest methods and in almost all cases SVM outperformed the others. This is the reason that motivated us to choose SVM as a baseline method while evaluating the performance of discriminative deep belief network (DDBN) in cancer classification.
In one of the few studies that utilized deep learning approach with microarray data, Gupta et al. [20] demonstrated the empirical effectiveness of using deep autoencoders as a pre-processing step for clustering of gene expression data. Another contribution was made by Fakoor at al. [21] that reported unsupervised feature learning can be used for cancer detection and cancer type analysis from gene expression data by deep stacked autoencoders. Ibrahim et al. [22] proposed a deep and active learning based method called MLFS (Multi-level gene/ MiRNA feature selection) for selecting genes from expression profiles. Their experiments show that the approach outperforms classical feature selection methods in hepatocellular carcinoma, lung cancer and breast cancer. In another study deep models are applied to functional genomic data to build low dimensional representations of multiple tracks of experimental functional genomics data [23]. Briefly, relevant literature studies that utilized deep learning approach in microarray data generally aimed at finding features or reducing dimensionality of the data in an unsupervised manner. This study diverges from them in that it aims at evaluating a deep learning method, namely Discriminative Deep Belief Network (DDBN), in supervised classification of cancer cases for the purpose of diagnosis.
While analyzing microarray gene expression data, a problem that should be handled is the high dimensionality of the data which causes the classifier to be overfitting to the data and increases the computational load. Even with limited number of samples in the data, there are thousands of features, i.e. genes. Therefore, prior to the classification, feature selection is essential for identifying the subset of genes which are relevant for predicting the classes of samples [24-27]. Additionally, imbalanced class distribution in the data is also another handicap for efficient training of a classifier, as well [28,29]. One way to deal with this problem is using oversampling techniques such as the Synthetic Minority Over-Sampling Technique (SMOTE) [30-32]. Blagus and Lusa [33] performed feature selection before using SMOTE in high dimensional gene expression data showed that substantial benefit can be obtained by use of SMOTE along with k-nearest neighbors classifier.
Being a recent approach, deep learning has not been investigated in application of classification of microarray gene expression data in the literature as comprehensively as it should have been. Therefore, research over the deep learning approach in this field is quite inadequate and as well as promising with the consideration of the fact that it is a proven method in other fields of machine learning [34-40]. In this study, our main goal is to show that DDBN is capable of being a successful decision support model for cancer data. For this purpose, three microarray gene expression datasets are analyzed: laryngeal cancer, bladder cancer and colorectal carcinomas. We compare the results of Discriminative Deep Belief Network (DDBN) with SVM in terms of accuracy, sensitivity, specificity, precision and F-measure metrics. In the preprocessing phase, we employed Information Gain (IG) feature selection method for discovering predictive genes, and SMOTE to overcome the problems aroused by the imbalanced nature of the data. Therefore, we propose a general model based on DDBN to diagnose cancer cases by using gene expression data. This model attempts to keep the diagnosis performance stable even with datasets containing imbalanced class distribution and high number of genes.
Materials and Methods
Description of the datasets
Microarray gene expression datasets of laryngeal cancer, bladder cancer and colorectal cancer are taken from BioGPS portal [41]. BioGPS is a customizable and extensible gene annotation portal and also a source for information about genes. It is supported by the U.S. National Institute of General Medical Sciences. It presents its gene annotation data to scientists by a flexible search interface. Additionally it has a role of content aggregator of many other gene annotation portals; therefore most of its content is obtained from other online resources.
Laryngeal microarray gene expression data was obtained using Affymetrix U133A Genechips. Tumor tissues of 66 laryngeal cancer patients were profiled and 22284 gene expressions are obtained. By adding age, duration of DFS (disease free survival) in months and the grade of the cancer a total of 22287 featured dataset is obtained.
Using Affymetrix U133 Plus 2.0 arrays platform bladder cancer gene expression data were obtained from exfoliated urothelia sampling for the evaluation of patients with suspected bladder cancer. The data was collected from 92 subjects and a total of 54676 genes are used. The aim of researchers was to identify urothelial cell transcriptomic signatures associated with bladder cancer.
The expression profiles of colorectal cancer were obtained from 89 patients using Affymetrix Human Genome U133 Plus 2.0 arrays. The aim of the researchers in collecting this data is to identify whether there is lymph node metastases in patient or not, because the existence of lymph node metastases give the physicians an idea about the prognosis of the colorectal cancer.
Table 1 summarizes the properties of three microarray gene expression data.
| Type of microarray gene expression data |
# of genes | Negatives | Positives | ||
|---|---|---|---|---|---|
| # of samples | description | # of samples | description | ||
| Laryngeal cancer [42] | 22284 | 75 | no recurrence of disease | 34 | recurrence of disease |
| Bladder cancer [43] | 54676 | 40 | non-cancer urothelial cells | 52 | urothelial cancer cells |
| Colorectal cancer [44] | 54676 | 57 | without lymph node metastasis | 32 | with lymph node metastasis |
Table 1. Class distributions and descriptions for microarray gene expression data.
Discriminative deep belief network (DDBN)
DDBN is a variant of Deep belief network (DBN) approach introduced by Hinton et al. [34]. A DBN is probabilistic generative model constructed by layers of Restricted Boltzman Machines (RBM). It can be trained by using Contrastive Divergence algorithm [34], in an unsupervised fashion. After completing the training of each layer, another RBM is concatenated as a new layer and trained by taking output of the previous layer as input. Required number of layers are obtained by adding one after another. This process is defined as greedy layer-wise learning method of DBN that provides an initialization for weights for network, in contrast to the traditional random initialization of neural networks.

Figure 1. A restricted boltzman machine.
As represented in Figure 1, an RBM consists of a layer of i binary visible units and j binary hidden units having bidirectional weighted connections. The energy of a configuration of this network can be defined by
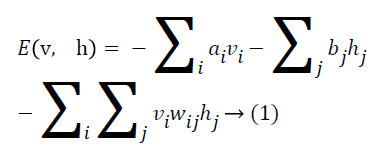
where ai is bias value of visible unit i, and bj is bias value of hidden unit j and wij is the weight between them. The probability of a visible vector v is
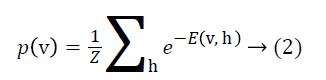
where Z is the normalizing factor calculated by summing all possible configurations of visible and hidden units. After observed variables are fed to visible units as input, a stochastic unit in a RBM has the probability of having value of 1
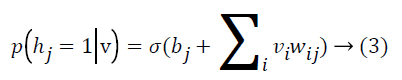
where σ(x)=1/(1+e-x) By using binary states of hidden units, the reconstructed binary states of visible units are
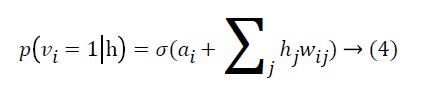
A DBN is obtained by a stack of RBMs, where the hidden layer of an RBM is the visible layer of the subsequent RBM. In the multilayered architecture of the DBN, there is a visible layer taking the input data to transmit it to the hidden layers. Each layer of the DBN is trained according to the training procedure of RBM. After training of RBM1 is completed, hidden units of RBM2 is added to the model. The hidden activations of RBM1 are fed to RBM2 as visible layer of RBM2 and the procedure is repeated for RBM3 and so on as represented in Figure 2A.

Figure 2. (a) A traditional DBN composed of three layers of RBM. (b) A DDBN composed of a visible unit, two RBM layers and an associative memory to output the target values.
If the model will be used as a discriminative model as represented in Figure 2B, the fine tuning procedure is performed by using back propagation algorithm. Firstly, a new layer which is label units of the data is added like the output layer of a neural network for producing the desired outputs, i.e. o1. Therefore, the weights of DDBN model are adjusted according to the difference of system output and actual expected values. In this model the last two layers are called associative memory for associating the lower layers to the label value. The only thing one should pay attention is to choose the correct learning rate. Because a large value of learning rate change the pre-determined weights a lot, and a small value causes a slow convergence.
Support vector machine (SVM)
SVM is a supervised machine learning method that depends on the statistical learning model proposed by Cortes and Vapnik [45] to use with classification and regression tasks. The statistical learning theory, which is also known as the Vapnik- Chernovenkis (VC) theory, analyses the problem of estimating the function from the training data. The models of statistical learning may be either parametric or nonparametric. SVM is a nonparametric model meaning that it has no assumptions on probability distributions of the input data and thus the model structure is not specified in advance.
In SVM classification, the inputs are defined as n-dimensional
feature vectors which are mapped to m-dimensional feature
space by kernel functions, where m>n. Inputs are classified
linearly by a hyperplane in such a higher dimensional space.
By using the training data, SVM learns a decision surface, i.e.
the hyperplane that separates the input vectors into two
different classes for binary classification. Therefore the aim of SVM classification is to design an optimum hyperplane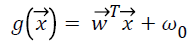 , where
, where 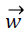 is vector of weights and
is vector of weights and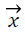 is the input vector. The vector determines the generalizing ability
of learning. There can be more than one solution in
determining the hyperplane but the best one leaves maximum
margin from both classes. This margin is determined according
to the closest data points called support vectors. For input
vectors from class 1 the hyperplane function produces values
larger than 1, and for input vectors from class 2 it produces values smaller than -1.
is the input vector. The vector determines the generalizing ability
of learning. There can be more than one solution in
determining the hyperplane but the best one leaves maximum
margin from both classes. This margin is determined according
to the closest data points called support vectors. For input
vectors from class 1 the hyperplane function produces values
larger than 1, and for input vectors from class 2 it produces values smaller than -1.
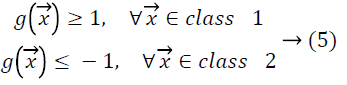
Let Z be the margin distance from support vectors of one of the classes to the hyperplane, then
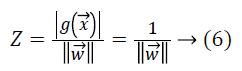
where we can see the total margin from both classes is 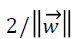 which must be maximized by minimizing the term
which must be maximized by minimizing the term 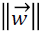 for
maximum separability. In case the two classes are nonseparable,
the amount proportional to number of misclassified
samples must also be minimized as in Equation 7,
for
maximum separability. In case the two classes are nonseparable,
the amount proportional to number of misclassified
samples must also be minimized as in Equation 7,
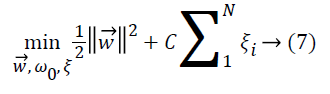
where ξi is the error parameter determined by input vectors that are not separated linearly and located on the wrong side of hyperplane. C is a predetermined constant parameter to control the penalty for misclassification. If this value is too small, many input vectors of misallocated are accepted, otherwise very few input vectors on wrong side are required.
The search for the optimum hyperplane in SVM is a nonlinear optimization problem solved by using Karush Kuhn Tucker conditions which use Lagrange multipliers i ≥ 0.
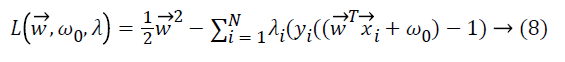
where yi is the corresponding class label of 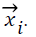
What makes the SVM approach being effective is the kernel functions that map the input vectors to feature vectors. By an appropriate mapping, data can be transformed to another dimensionality in which it can be separated by a hyperplane. Quadratic, polynomial, sigmoid, linear and radial basis functions are some common kernel functions.
Information gain for gene selection
Information Gain (IG) measures the reduction in uncertainty about a class variable, therefore it is entropy based filter method. Formally, assume Y is the class attribute of a data set and X is a given feature, i.e. gene. The IG of X is the reduction of uncertainty of Y values when X values are known. This uncertainty is measured as H(Y), the entropy of Y. IG of X is the difference between entropy of Y and entropy of Y after X values are observed, and is calculated as in Equation 9.
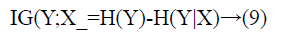
Entropy of Y is calculated with Equation 10.
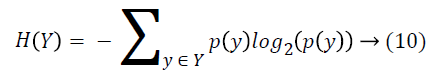
where y is a value of Y class feature, and p(y) is the probability of Y=y. And entropy of Y after X values are observed is calculated as:
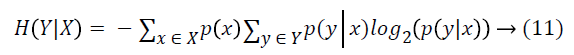
Synthetic minority over-sampling technique (SMOTE)
Chawla et al. [30] proposed the SMOTE which resolves the imbalanced class distribution problem by oversampling minority class instances in the data. In classification tasks, the problem arises when the classifier biases towards a majority class when learning from an imbalanced data. In the literature, it is shown that classifiers are better in balanced datasets compared to their performances in imbalanced ones [31,32]. SMOTE creates new instances from a minor class based on the idea that closest vectors have the same class value. Each instance is considered as a vector, and the closest vectors are determined by k-nearest neighbors algorithm. A new sample is generated along the line between the minority sample and a neighbor selected from k-nearest neighbors randomly. A difference vector is calculated by taking the difference between the minority class sample x and the neighbor sample xn. Finally, a new synthetic sample vector xnew is generated by:
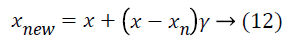
where γ is a random value between 0 and 1. The output of algorithm is the synthetic samples that count to a percent of number of minority samples which is a parameter the algorithm takes. In the case of microarray gene expression data classification, especially laryngeal and colorectal data are imbalanced. However SMOTE is applied to all three data, the aim is to minimize the potential misclassification caused by an unbalanced structure of data.
Results and Discussion
In order to measure the classification performance of the deep learning approach, experiments are conducted over the three datasets mentioned previously. The metrics of accuracy, sensitivity, specificity, precision and F-measure are used to evaluate the classification capacity. All these experiments were carried out using the ten-folds cross validation technique. By this technique the model is validated by taking the average of ten tests each of which is executed by using one out of ten subsets of the dataset for testing and the remaining parts for training. SVM is employed with polynomial kernel which is determined according to comparative pre-studies on different kernels. As for DBN, we used a two layered structure with 50 and 10 hidden nodes in the first and second hidden layers, respectively. Training of the network is achieved with a learning rate of 0.04 and a momentum of 0.9.
Before the deep analysis of DDBN and SVM comparison, Table 2 is presented that enables to comprehend the reason of selection of methods. In Table 2 the methods of Random Forest and k-Nearest Neighbors are included besides DDBN and SVM. The values are accuracies of classification of each method after application of IG and SMOTE preprocessing steps. According to Table 2, Random Forest generates considerable results, but in a total assessment performance values of DDBN and SVM are better than Random Forest and k-NN.
| Data | DDBN | Random Forest | SVM | k-NN |
|---|---|---|---|---|
| Laryngeal | 0.944 | 90.21 | 0.937 | 73.43 |
| Bladder | 0.947 | 92.42 | 0.962 | 87.12 |
| Colorectal | 0.909 | 87.60 | 0.826 | 78.51 |
Table 2. Accuracies of classification of four methods.
Table 3, Table 4 and Table 5 summarize the classification results of DDBN and SVM for laryngeal, bladder and colorectal microarray gene expression data respectively. In each of the tables, results are grouped into four categories. First category is the no preprocessing (no prep.) category in which each data is directly classified by DDBN and SVM without any preprocessing applied to the data. The second category represents the results of classification of DDBN and SVM after selecting discriminative genes of cancer data by the IG filter. As IG produces a score for each feature, we took the top ranking features exceeding 0.1 threshold score, therefore for laryngeal, bladder and colorectal microarray gene expression data, 251, 300 and 322 features are used, respectively. In the third category, the results are based on SMOTE based preprocessing while the last category presents the results obtained by applying both IG and SMOTE subsequently. The SMOTE algorithm produces new synthetic instances whose amount is determined by an algorithm parameter. This parameter is the percent of number of minority samples to be considered while generating new samples. In the study, this parameter is set to 100% and therefore all minority samples are used for new samples. As a result, the number of minor samples are doubled. Another parameter of SMOTE is the number of nearest neighbors which is determined as 5.
| Methods | Accuracy | Sensitivity | Specificity | Precision | F-measure | |
|---|---|---|---|---|---|---|
| DDBN | No prep. | 0.716 | 0.500 | 0.813 | 0.548 | 0.523 |
| IG | 0.881 | 0.853 | 0.893 | 0.784 | 0.817 | |
| SMOTE | 0.860 | 0.941 | 0.787 | 0.800 | 0.865 | |
| IG + SMOTE | 0.944 | 0.985 | 0.907 | 0.905 | 0.943 | |
| SVM | No prep. | 0.569 | 0.294 | 0.693 | 0.303 | 0.299 |
| IG | 0.872 | 0.794 | 0.907 | 0.794 | 0.794 | |
| SMOTE | 0.832 | 0.971 | 0.707 | 0.75 | 0.846 | |
| IG + SMOTE | 0.937 | 0.956 | 0.920 | 0.915 | 0.935 | |
Table 3. Results of evaluation of laryngeal microarray gene expression data using DDBN and SVM (with polynomial kernel).
| Methods | Accuracy | Sensitivity | Specificity | Precision | F-measure | |
|---|---|---|---|---|---|---|
| DDBN | No prep. | 0.533 | 0.712 | 0.300 | 0.569 | 0.633 |
| IG | 0.913 | 0.942 | 0.875 | 0.907 | 0.950 | |
| SMOTE | 0.818 | 0.558 | 0.988 | 0.967 | 0.708 | |
| IG + SMOTE | 0.947 | 0.865 | 1.000 | 1.000 | 0.928 | |
| SVM | No prep. | 0.707 | 0.808 | 0.575 | 0.712 | 0.757 |
| IG | 0.946 | 0.923 | 0.975 | 0.980 | 0.950 | |
| SMOTE | 0.909 | 0.808 | 0.975 | 0.855 | 0.875 | |
| IG + SMOTE | 0.962 | 0.904 | 1.000 | 1.000 | 0.949 | |
Table 4. Results of evaluation of bladder microarray gene expression data using DDBN and SVM (with polynomial kernel).
| Methods | Accuracy | Sensitivity | Specificity | Precision | F-measure | |
|---|---|---|---|---|---|---|
| DDBN | No prep. | 0.640 | 0.188 | 0.895 | 0.500 | 0.273 |
| IG | 0.798 | 0.594 | 0.912 | 0.792 | 0.679 | |
| SMOTE | 0.471 | 0.750 | 0.158 | 0.500 | 0.600 | |
| IG + SMOTE | 0.909 | 1.000 | 0.807 | 0.853 | 0.921 | |
| SVM | No prep. | 0.640 | 0.313 | 0.312 | 0.500 | 0.385 |
| IG | 0.719 | 0.563 | 0.807 | 0.621 | 0.590 | |
| SMOTE | 0.868 | 0.984 | 0.737 | 0.808 | 0.887 | |
| IG + SMOTE | 0.826 | 0.891 | 0.754 | 0.803 | 0.844 | |
Table 5. Results of evaluation of colorectal microarray gene expression data using DDBN and SVM (with polynomial kernel).
In Figure 3, it can be seen that the application of both preprocessing steps of IG and SMOTE contributed to more stable results. In order to recognize DDBN or SVM to be successful the result in each metric should approximate to 1. Therefore we expect each curve would be as flat as possible in the alignment of 1 score.

Figure 3. Comparison of DDBN vs. SVM in (a) laryngeal cancer (b) bladder cancer (c) colorectal cancer datasets. The effect of SMOTE and IG preprocessing are also given for each dataset.
In Figures 3A and 3B the results of laryngeal and bladder carcinoma after application of IG and SMOTE are comparable for DDBN and SVM. In Figure 3C, performance of DDBN on bladder carcinoma without preprocessing is very low in sensitivity while it is very high in specificity which caused instability in the curve. This is because the major of the bladder data is classified to negatives and thus positive instances are classified incorrectly i.e., very few instances are decided to be positive. In such cases, F-measure is more reliable than using sensitivity and specificity alone to measure the overall performance, since F-measure weighs positive and negative cases in a more balanced way. Although DDBN and SVM seem to produce nearly the same accuracy in colorectal carcinoma data in no preprocessing category, SVM is more stable than DDBN in both detecting positive and negative cases. On colorectal gene expression data, the application of IG and SMOTE provided a considerable contribution to the improvement of results, especially to the results of DDBN. It should be noted that in classification tasks the performance results are very dependent on the data and in this context colorectal carcinoma data is observed to be more sensitive to preprocessing steps. This is because this dataset suffers more from the imbalanced class distribution problem with relatively less number of samples in the dataset. In this dataset with the application of IG and SMOTE, DDBN has outperformed SVM by accuracy, sensitivity and specificity values of 0.909, 1.000 and 0.807, respectively. Considering all the three datasets, Table 6 shows that in terms of average values DDBN has outperformed SVM in all metrics.
| Evaluation metrics | Averages of results | |
|---|---|---|
| DDBN | SVM | |
| Accuracy | 0.933 | 0.908 |
| Sensitivity | 0.950 | 0.917 |
| Specificity | 0.905 | 0.891 |
| Precision | 0.912 | 0.906 |
| F-measure | 0.931 | 0.909 |
Table 6. Averages of results in each metric from three microarray gene expression data after preprocessing of IG and SMOTE.
Using t-test on the difference of the corresponding accuracy values of DDBN and SVM, the value of 0.842 is obtained on the t-distribution with 2 degrees of freedom. In this case the obtained p-value is greater than 0.10. H0 hypothesis is not rejected which indicates the mean of the differences of the accuracies is 0. Therefore according to the analysis of the results there is not statistical significance between DDBN and SVM. This verifies that classification performance of DDBN is at least as good as that of SVM.
In the first dataset where imbalanced class distribution is the most obvious, number of negatives are more than the double of number of positives, DDBN clearly outperforms SVM without any preprocessing. Another distinct feature of the dataset is its number of features, being nearly half of features in other datasets. In this dataset, the performance of SVM can only be comparable to that of DDBN with preprocessing. In the other datasets, the second and the third, the class imbalance problem is slighter when compared to the first dataset. In the second dataset SVM is better without any preprocessing while the final results of the two algorithms with preprocessing are comparable. In the third one, the performances are similar without preprocessing but DDBN clearly outperforms SVM with preprocessing. Since the structural features of these two datasets are similar, different performances of DDBN and SVM suggest that their performances may vary depending on the dataset. However, in overall performance, DDBN appeared to perform better in terms of the F-measure metric which alone can safely be used to compare the overall classification performance of a classifier [46].
As a result, empirical results of the study suggest that DDBN has the potential as a decision support system in gene expression data based cancer diagnosis. The layered structure of DDBN is convenient for identifying relevant genes in microarray gene expression data. Each layer produces the features of their input feature values and this leaded to more success of DDBN in high dimensional data. As a deep model, DDBN tends to extract a relevant set of features from the input layer and then it enhances the recognition process in the next layer. The working principle of the presented network structure allows learning the input statistics in the unsupervised greedy layer wise training phase. This knowledge is then used in the fine tuning phase by back propagation algorithm.
With the proposed model that used IG and SMOTE preprocessing steps, the DDBN algorithm can be generalized to perform adequately in other datasets of cancer, as well. By use of preprocessing, the processing time is reduced and the performance can be improved. According to Table 6 the overall diagnosis performance of DDBN is 93.3% accuracy, considering the datasets of this study which appeared to be adequate for most of the cases.
Conclusions
In this study we adapted a deep learning approach, namely DDBN to the problem of gene expression data classification. The method includes a preprocessing phase with IG and SMOTE and a classification phase with DDBN. The effectiveness of the method is demonstrated by using laryngeal, bladder and colorectal microarray gene expression datasets. The results of DDBN are compared with those of SVM which is proven to be very successful in microarray gene expression classification in recent studies [14-18].
The experiments showed that in microarray gene expression data analysis both DDBN and SVM achieved promising results after eliminating the effect of redundant and inconsistent features by use of preprocessing steps of IG and SMOTE. In average values, DDBN outperformed SVM in all metrics. For future studies other deep learning methods besides DDBN are worth investigating for not only other types of cancers but also in different areas of biomedicine.
Acknowledgement
This study was financially supported by the Cukurova University Research Foundation (Project No: FDK-2015-4395).
References
- US Cancer Statistics Working Group. United States cancer statistics: 1999-2010 incidence and mortality web-based report. Atlanta: US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute 2013.
- Su Z, Hong H, Perkins R, Shao X, Cai W, Tong W. Consensus analysis of multiple classifiers using non-repetitive variables: diagnostic application to microarray gene expression data. Comput Biol Chem 2007; 31: 48-56.
- Lu Y, Han J. Cancer classification using gene expression data. Informa Syst 2003; 28: 243-268.
- Tarca AL, Romero R, Draghici S. Analysis of microarray experiments of gene expression profiling. Am J Obstetrics Gynecol 2006; 195: 373-388.
- Senthilkumar B, Umamaheswari G. Combination of Novel Enhancement Technique and Fuzzy C Means Clustering Technique in Breast Cancer Detection. Biomed Res 2013; 24: 252-256.
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh M, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999; 286: 531-537.
- Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, Meltzer PS. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 2001; 7: 673-679.
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Bordrick JC, Sabet H,Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Lu L, Lwis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R,Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lyumphoma identified by gene expression profiling. Nature 2000; 403: 503-511.
- Samarasinghe S, Chaiboonchoe A. Neural Networks and Fuzzy Clustering Methods for Assessing the Efficacy of Microarray Based Intrinsic Gene Signatures in Breast Cancer Classification and the Character and Relations of Identified Subtypes. Artific Neural Net 2015; 285-317.
- Dudoit S, Fridlyand J, Speed P. Comparison of discrimination methods for classification of tumors using gene expression data. J Am Stat Assoc 2002; 97: 77-87.
- Lee JW, Lee JB, Park M, Song SH. An extensive comparison of recent classification tools applied to microarray data. Comput Stat Data Anal 2005; 48: 869-885.
- Vural H, Subasi A. Data-Mining Techniques to Classify Microarray Gene Expression Data Using Gene Selection by SVD and Information Gain. Model Artificial Intel 2015; 6: 171-182.
- Yang S, Naiman DQ. Multiclass cancer classification based on gene expression comparison. Stat Appl Genetic Mol Biol 2014; 13: 477-496.
- Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics 2000; 16: 906-914.
- Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M, Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Nat Acad Sci 2000; 97: 262-267.
- Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov JP, Poggio T, Gerald W, Loda M, Lander ES, Golub TR. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Nat Acad Sci 2001; 98: 15149-15154.
- Mukherjee S. Classifying microarray data using support vector machines. A practical approach to microarray data analysis 2003; 1: 166-185.
- Statnikov A, Aliferis CF, Tsamardinos I, Hardin D, Levy S. A comprehensive evaluation of multicategory classification methods for microarray gene expression cancer diagnosis. Bioinformatics 2005; 21: 631-643.
- Pirooznia M, Yang JY, Yang MQ, Deng Y. A comparative study of different machine learning methods on microarray gene expression data. BMC Genomics 2008; 9: 1.
- Gupta A, Wang H, Ganapathiraju M. Learning structure in gene expression data using deep architectures, with an application to gene clustering. In Bioinformatics and Biomedicine (BIBM), 2015 IEEE International Conference on, pp. 1328-1335, IEEE.
- Fakoor R, Ladhak F, Nazi A, Huber M. Using deep learning to enhance cancer diagnosis and classification. In Proceedings of the ICML Workshop on the Role of Machine Learning in Transforming Healthcare. Atlanta, Georgia: JMLR: W&CP.
- Ibrahim R, Yousri NA, Ismail MA, El-Makky NM. Multi-level gene/MiRNA feature selection using deep belief nets and active learning. In 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society 2014; pp. 3957-3960. IEEE.
- Denas O. Deep Models for Gene Regulation. Diss. Emory University, 2014.
- Kumar AP, Preeja V. Feature Selection for high Dimensional DNA Microarray data using hybrid approaches. Bioinformation 2013; 9: 824.
- Bolón-Canedo V, Sánchez-Maroño N, Alonso-Betanzos A. Distributed feature selection: An application to microarray data classification. Appl Soft Comput 2015; 30: 136-150.
- Fernández-Navarro F, Hervás-Martínez C, Ruiz R, Riquelme JC. Evolutionary generalized radial basis function neural networks for improving prediction accuracy in gene classification using feature selection. Appl Soft Comput 2012; 12: 1787-1800.
- Chen Y, Zhao Y. A novel ensemble of classifiers for microarray data classification. Appl Soft Comput 2008; 8: 1664-1669.
- Kamal AH, Zhu X, Pandya AS, Hsu S, Shoaib M. The impact of gene selection on imbalanced microarray expression data. In Bioinformatics and Computational Biology 2009; pp. 259-269. Springer Berlin Heidelberg.
- Yu H, Ni J, Zhao J. ACOSampling: An ant colony optimization-based undersampling method for classifying imbalanced DNA microarray data. Neurocomputing 2013; 101: 309-318.
- Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. SMOTE: synthetic minority over-sampling technique. J Artif Intel Res 2002; 321-357.
- Chawla NV. C4. 5 and imbalanced data sets: investigating the effect of sampling method, probabilistic estimate, and decision tree structure. Proceedings of the ICML, 2003.
- Estabrooks A, Jo T, Japkowicz N. A multiple resampling method for learning from imbalanced data sets. Comput Intel 2004; 20: 18-36.
- Blagus R, Lusa L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinforma 2013; 14: 106.
- Hinton GE, Osindero S, Teh YW. A fast learning algorithm for deep belief nets. Neural computation 2006; 18: 1527-1554.
- Mohamed A, Dahl GE, Hinton G. Acoustic modeling using deep belief networks. IEEE Transact Audio Speech Lang Process 2012; 20: 14-22.
- Vinyals O, Ravuri SV. Comparing multilayer perceptron to Deep Belief Network Tandem features for robust ASR. In: Acoustics, Speech and Signal Processing (ICASSP), 2011 IEEE International Conference on. IEEE, 2011; p. 4596-4599.
- Bengio Y, Lamblin P, Popovici D, Larochelle H. Greedy layer-wise training of deep networks. Advances in Neural Information Processing Systems 2007; 19: 153.
- Sarikaya R, Hinton GE, Ramabhadran B. Deep belief nets for natural language call-routing. Acoustics, Speech and Signal Processing, 2011 IEEE International Conference on. IEEE.
- Huang GB, Lee H, Learned-Miller E. Learning hierarchical representations for face verification with convolutional deep belief networks. In Computer Vision and Pattern Recognition (CVPR), 2012 IEEE Conference on, pp. 2518-2525, IEEE.
- Sainath TN, Kingsbury B, Ramabhadran B, Fousek P, Novak P, Mohamed AR. Making deep belief networks effective for large vocabulary continuous speech recognition. In Automatic Speech Recognition and Understanding, 2011 IEEE Workshop on, pp. 30-35, IEEE.
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW 3rd, Su AI. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009; 10:R130.
- Fountzilas E, Kotoula V, Angouridakis N, Karasmanis I, Wirtz RM, Eleftheraki AG, Veltrup E, Markou K, Nikolaou A, Pectasides D, Fountzilas G. Identification and validation of a multigene predictor of recurrence in primary laryngeal cancer. PloS one 2013; 8: e70429.
- Urquidi V, Goodison S, Cai Y, Sun Y, Rosser CJ. A candidate molecular biomarker panel for the detection of bladder cancer. Cancer Epidemiol Biomark Prevent 2012; 21: 2149-2158.
- Watanabe T, Kobunai T, Tanaka T, Ishihara S, Matsuda K, Nagawa H. Gene expression signature and the prediction of lymph node metastasis in colorectal cancer by DNA microarray. Dise Colon Rectum 2009; 52: 1941-1948.
- Cortes C, Vapnik V. Support-vector networks. Machine learning 1995; 20: 273-297.
- Tang Y, Zhang YQ, Chawla NV, Krasser S. SVMs modeling for highly imbalanced classification. Systems, Man, and Cybernetics, Part B: Cybernetics, IEEE Transactions on 2009; 39: 281-288.